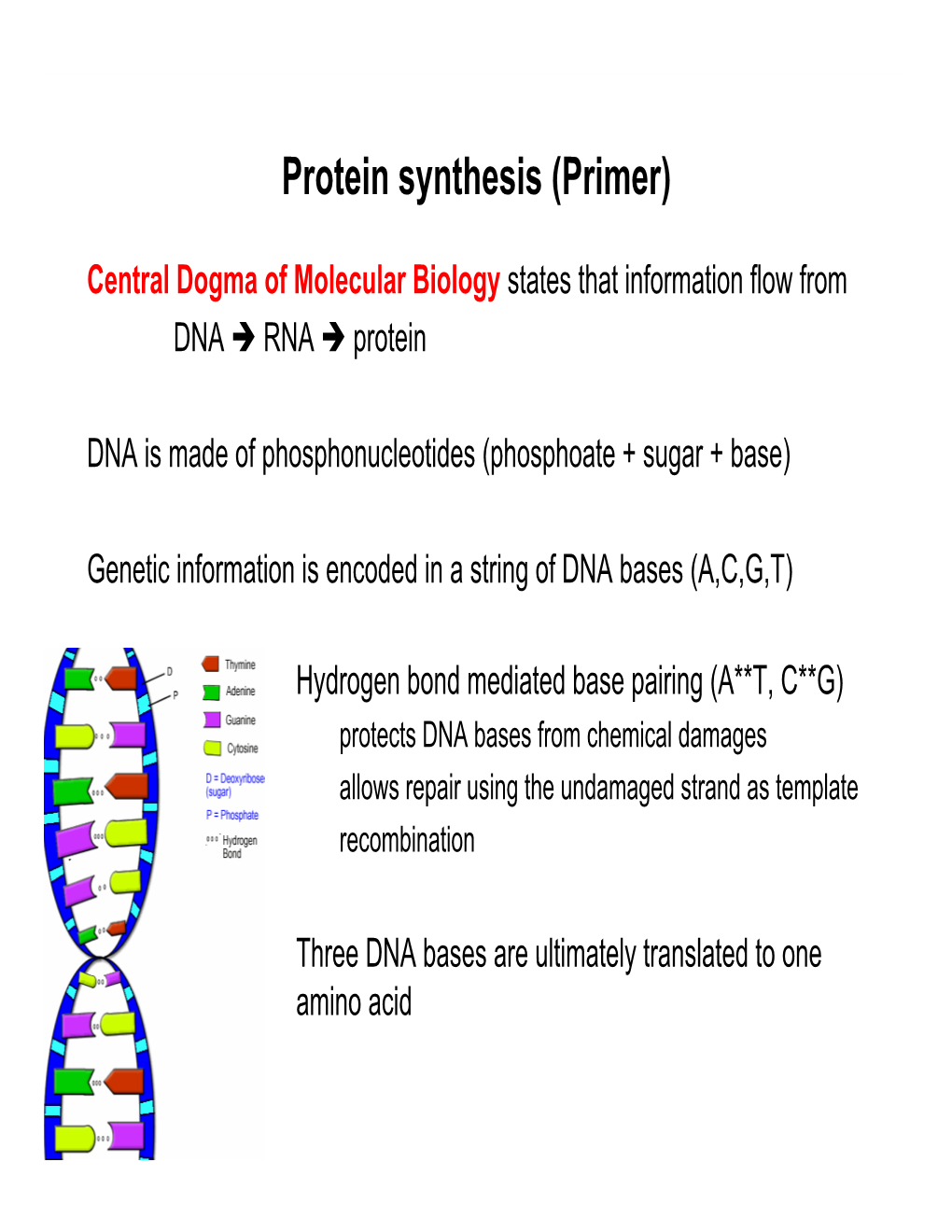
Protein Synthesis (Primer)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Simulations of Я-Hairpin Folding Confined to Spherical Pores Using
Simulations of -hairpin folding confined to spherical pores using distributed computing D. K. Klimov*†, D. Newfield‡, and D. Thirumalai*† *Institute for Physical Science and Technology, University of Maryland, College Park, MD 20742; and ‡Parabon Computation, 3930 Walnut Street, Suite 100, Fairfax, VA 22030-4738 Communicated by George H. Lorimer, University of Maryland, College Park, MD, April 12, 2002 (received for review December 18, 2001) 3 ϱ We report the thermodynamics and kinetics of an off-lattice Go radius of gyration of a chain Rg at D (the size of a chain in  Ӎ model -hairpin from Ig-binding protein confined to an inert bulk solution) to N according to Rg aN . If the chain is ideal, ϭ ⌬ ϭ ͞ 2 spherical pore. Confinement enhances the stability of the hairpin then 0.5 and FU RTN(a D) . Because of the reduction due to the decrease in the entropy of the unfolded state. Compared in the translational entropy, confinement also increases the free ⌬ Ͼ ⌬ ͞⌬ ϽϽ with their values in the bulk, the rates of hairpin formation increase energy of the native state, i.e., FN 0. If FN FU 1, then in the spherical pore. Surprisingly, the dependence of the rates on localization of a protein in a confined space stabilizes the native the pore radius, Rs, is nonmonotonic. The rates reach a maximum state compared with the bulk. It also follows that there is a range ͞ b Ӎ b at Rs Rg,N 1.5, where Rg,N is the radius of gyration of the folded of D values over which stability is maximized. -

The Structure of Small Beta Barrels
bioRxiv preprint doi: https://doi.org/10.1101/140376; this version posted May 24, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. The Structure of Small Beta Barrels Philippe Youkharibache*, Stella Veretnik1, Qingliang Li, Philip E. Bourne*1 National Center for Biotechnology Information, The National Library of Medicine, The National Institutes of Health, Bethesda Maryland 20894 USA. *To whom correspondence should be addressed at [email protected] and [email protected] 1 Current address: Department of Biomedical Engineering, The University of Virginia, Charlottesville VA 22908 USA. 1 bioRxiv preprint doi: https://doi.org/10.1101/140376; this version posted May 24, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Abstract The small beta barrel is a protein structural domain, highly conserved throughout evolution and hence exhibits a broad diversity of functions. Here we undertake a comprehensive review of the structural features of this domain. We begin with what characterizes the structure and the variable nomenclature that has been used to describe it. We then go on to explore the anatomy of the structure and how functional diversity is achieved, including through establishing a variety of multimeric states, which, if misformed, contribute to disease states. -

Inclusion of a Furin Cleavage Site Enhances Antitumor Efficacy
toxins Article Inclusion of a Furin Cleavage Site Enhances Antitumor Efficacy against Colorectal Cancer Cells of Ribotoxin α-Sarcin- or RNase T1-Based Immunotoxins Javier Ruiz-de-la-Herrán 1, Jaime Tomé-Amat 1,2 , Rodrigo Lázaro-Gorines 1, José G. Gavilanes 1 and Javier Lacadena 1,* 1 Departamento de Bioquímica y Biología Molecular, Facultad de Ciencias Químicas, Universidad Complutense de Madrid, Madrid 28040, Spain; [email protected] (J.R.-d.-l.-H.); [email protected] (J.T.-A.); [email protected] (R.L.-G.); [email protected] (J.G.G.) 2 Centre for Plant Biotechnology and Genomics (UPM-INIA), Universidad Politécnica de Madrid, Pozuelo de Alarcón, Madrid 28223, Spain * Correspondence: [email protected]; Tel.: +34-91-394-4266 Received: 3 September 2019; Accepted: 10 October 2019; Published: 12 October 2019 Abstract: Immunotoxins are chimeric molecules that combine the specificity of an antibody to recognize and bind tumor antigens with the potency of the enzymatic activity of a toxin, thus, promoting the death of target cells. Among them, RNases-based immunotoxins have arisen as promising antitumor therapeutic agents. In this work, we describe the production and purification of two new immunoconjugates, based on RNase T1 and the fungal ribotoxin α-sarcin, with optimized properties for tumor treatment due to the inclusion of a furin cleavage site. Circular dichroism spectroscopy, ribonucleolytic activity studies, flow cytometry, fluorescence microscopy, and cell viability assays were carried out for structural and in vitro functional characterization. Our results confirm the enhanced antitumor efficiency showed by these furin-immunotoxin variants as a result of an improved release of their toxic domain to the cytosol, favoring the accessibility of both ribonucleases to their substrates. -

Protein Structure Databases and Classification
Protein Structure Databases and Classification •SCOP, CATH classification schemes, what they mean. •Motifs: classic turn types. Extended turn types. •TOPS: drawing a protein molecule The SCOP database • Contains information about classification of protein structures and within that classification, their sequences • Go to http://scop.berkeley.edu SCOP classification heirarchy global characteristics (no (1) class evolutionary relation) (2) fold Similar “topology” . Distant (3) superfamily evolutionary cousins? (4) family Clear structural homology (5) protein Clear sequence homology (6) species functionally identical unique sequences protein classes 1. all α (126) number of sub-categories 2. all β (81) 3. α/β (87) 4. α+β (151) 5. multidomain (21) 6. membrane (21) 7. small (10) 8. coiled coil (4) 9. low-resolution (4) possibly not complete, or 10. peptides (61) erroneous 11. designed proteins (17) class: α/β proteins Mainly parallel beta sheets (beta-alpha-beta units) Folds: TIM-barrel (22) swivelling beta/beta/alpha domain (5) spoIIaa-like (2) flavodoxin-like (10) restriction endonuclease-like (2) ribokinase-like (2) Many folds have historical names. chelatase-like (2) “TIM” barrel was first seen in TIM. These classifications are done by eye, mostly. fold: flavodoxin-like 3 layers, α/β/α; parallel beta-sheet of 5 strand, order 21345 Superfamilies: 1.Catalase, C-terminal domain (1) Note the term: “layers” 2.CheY-like (1) 3.Succinyl-CoA synthetase domains (1) These are not domains. 4.Flavoproteins (3) No implication of 5.Cobalamin (vitamin B12)-binding domain (1) structural independence. 6.Ornithine decarboxylase N-terminal "wing" domain (1) Note how beta sheets are 7.Cutinase-like (1) described: number of 8.Esterase/acetylhydrolase (2) strands, order (N->C) 9.Formate/glycerate dehydrogenase catalytic domain-like (3) 10.Type II 3-dehydroquinate dehydratase (1) fold-level similarity common topological features catalase flavodoxin At the fold level, a common core of secondary structure is conserved. -

Β-Barrel Oligomers As Common Intermediates of Peptides Self
www.nature.com/scientificreports OPEN β-barrel Oligomers as Common Intermediates of Peptides Self-Assembling into Cross-β Received: 20 April 2018 Accepted: 22 June 2018 Aggregates Published: xx xx xxxx Yunxiang Sun, Xinwei Ge, Yanting Xing, Bo Wang & Feng Ding Oligomers populated during the early amyloid aggregation process are more toxic than mature fbrils, but pinpointing the exact toxic species among highly dynamic and heterogeneous aggregation intermediates remains a major challenge. β-barrel oligomers, structurally-determined recently for a slow-aggregating peptide derived from αB crystallin, are attractive candidates for exerting amyloid toxicity due to their well-defned structures as therapeutic targets and compatibility to the “amyloid- pore” hypothesis of toxicity. To assess whether β-barrel oligomers are common intermediates to amyloid peptides - a necessary step toward associating β-barrel oligomers with general amyloid cytotoxicity, we computationally studied the oligomerization and fbrillization dynamics of seven well- studied fragments of amyloidogenic proteins with diferent experimentally-determined aggregation morphologies and cytotoxicity. In our molecular dynamics simulations, β-barrel oligomers were only observed in fve peptides self-assembling into the characteristic cross-β aggregates, but not the other two that formed polymorphic β-rich aggregates as reported experimentally. Interestingly, the latter two peptides were previously found nontoxic. Hence, the observed correlation between β-barrel oligomers formation and cytotoxicity supports the hypothesis of β-barrel oligomers as the common toxic intermediates of amyloid aggregation. Aggregation of proteins and peptides into amyloid fbrils is associated with more than 25 degenerative diseases, including Alzheimer’s disease (AD)1,2, Parkinson’s disease (PD)3,4, prion conditions5 and type-2 diabetes (T2D)6,7. -

Predicting Protein Secondary and Supersecondary Structure
29 Predicting Protein Secondary and Supersecondary Structure 29.1 Introduction............................................ 29-1 Background • Difficulty of general protein structure prediction • A bottom-up approach 29.2 Secondary structure ................................... 29-5 Early approaches • Incorporating local dependencies • Exploiting evolutionary information • Recent developments and conclusions 29.3 Tight turns ............................................. 29-13 29.4 Beta hairpins........................................... 29-15 29.5 Coiled coils ............................................. 29-16 Early approaches • Incorporating local dependencies • Predicting oligomerization • Structure-based predictions • Predicting coiled-coil protein interactions Mona Singh • Promising future directions Princeton University 29.6 Conclusions ............................................ 29-23 29.1 Introduction Proteins play a key role in almost all biological processes. They take part in, for example, maintaining the structural integrity of the cell, transport and storage of small molecules, catalysis, regulation, signaling and the immune system. Linear protein molecules fold up into specific three-dimensional structures, and their functional properties depend intricately upon their structures. As a result, there has been much effort, both experimental and computational, in determining protein structures. Protein structures are determined experimentally using either x-ray crystallography or nuclear magnetic resonance (NMR) spectroscopy. While -

JBB2026 Fall 2018 Gil Privé Protein Structure • Peptide Conformations
JBB2026 Fall 2018 Gil Privé Protein Structure • peptide conformations and residue preferences • elements of secondary structure • supersecondary structure and motifs • packing of helices and sheets • chain topologies • internal packing • protein interfaces • membrane proteins • multimeric proteins • domain motions The Machinery of Life David S. Goodsell http://mgl.scripps.edu/people/goodsell Figure 1. Transcription and RNA processing in the nucleus. Figure 2. Transport through the nuclear pore. Figure 3. Endoplasmic reticulum. Figure 4. Transport from the endoplasmic reticulum. Figure 5. Protein sorting in the Golgi. Plasma cell - IgG secretion Figure 6. Transport from the Golgi. Figure 7. Transport of a vesicle through the cytoplasm. David Goodsell The Machinery of Life Figure 8. Export of proteins across the cell membrane. http://www.3dmoleculardesigns.com/Teacher-Resources/Tour-of-a-Human-Cell.htm Eukaryotic cell panorama 1. Transcription and RNA processing in the nucleus. 2. Transport through the nuclear pore. Biochemistry and Molecular Biology Education Yellow: DNA, proteins Volume 39, Issue 2, pages 91-101, 28 MAR 2011 DOI: 10.1002/bmb.20494 Pink: RNA, proteins http://onlinelibrary.wiley.com/doi/10.1002/bmb.20494/full#fig2 Blue: Cytoplasmic proteins http://www.3dmoleculardesigns.com/Teacher-Resources/Tour-of-a-Human-Cell.htm Purple: Ribosomes Green: Membranes,proteins 1. Transcription and RNA 2. Transport through the 3. Endoplasmic reticulum. processing in the nucleus. nuclear pore. 4. Transport from the endoplasmic reticulum. 5. Protein sorting in the golgi. 6. Transport from the golgi. 7. Transport of a vesicle 8. Export of proteins across the through the cytoplasm. cell membrane. Tyr Thr Gly Cys Ile Ile Ala Gly The structure (conformer) defined by the dihedral angles main chain (φ,ψ) side chains (!1, !2, …) φ =180 ; ψ=180 φ =-60 ; ψ=-45 φ ψ ω φ ψ Note: unsaturated C-N bond length is 1.45 Å -Peptide bond has ~40% double bond character - dihedral is constrained. -

Beta Structure Motifs of Islet Amyloid Polypeptides Identified Through Surface-Mediated Assemblies
Beta structure motifs of islet amyloid polypeptides identified through surface-mediated assemblies Xiao-Bo Mao (毛晓波)a,1, Chen-Xuan Wang (王晨轩)a,b,1, Xing-Kui Wu (吴兴奎)a, Xiao-Jing Ma (马晓晶)a,c, Lei Liu (刘磊)a,LanZhang(张岚)a,LinNiu(牛琳)a, Yuan-Yuan Guo (郭元元)a,Deng-HuaLi(李灯华)a, Yan-Lian Yang (杨延莲)a,2, and Chen Wang (王琛)a,2 aNational Center for Nanoscience and Technology, Beijing 100190, China; bDepartment of Chemistry, Tsinghua University, Beijing 100084, China; and cState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Science, Changchun 130022, China Edited by* David Eisenberg, University of California, Los Angeles, CA, and approved September 29, 2011 (received for review February 22, 2011) We report here the identification of the key sites for the beta struc- been made in the aggregation behaviors of the mutant analogs ture motifs of the islet amyloid polypeptide (IAPP) analogs by using of IAPP (2, 7, 12, 15, 21–24), while more evidence is still needed scanning tunneling microscopy (STM). Duplex folding structures in to gain insight into the relationship between primary sequences of human IAPP8–37 (hIAPP8–37) assembly were observed featuring the IAPP analogs and the conformational variants. a hairpin structure. The multiplicity in rIAPP assembly structures It is generally acknowledged that the noncrystalline nature of indicates the polydispersity of the rat IAPP8–37 (rIAPP8–37) beta-like the fibril structures formed by amyloidal peptides makes it experi- motifs. The bimodal length distribution of beta structure motifs mentally challenging for structural analyses at molecular level. -

Peptide Folding Simulations S Gnanakaran, Hugh Nymeyer, John Portman, Kevin Y Sanbonmatsu and Angel E Garcı´A
Peptide folding simulations S Gnanakaran, Hugh Nymeyer, John Portman, Kevin Y Sanbonmatsu and Angel E Garcı´aà Developments in the design of small peptides that mimic urational entropy, hydrogen bond formation, solvation, proteins in complexity, recent advances in nanosecond hydrophobic core formation and ion pair formation deter- time-resolved spectroscopy methods to study peptides and the mines the folding rate and stability of proteins. This development of modern, highly parallel simulation algorithms competition plays an essential role throughout the fold- have come together to give us a detailed picture of peptide ing process and determines the thermodynamic equili- folding dynamics. Two newly implemented simulation brium between folded and unfolded states. Modeling techniques, parallel replica dynamics and replica exchange this competition is a standing challenge in peptide fold- molecular dynamics, can now describe directly from simulations ing simulations. the kinetics and thermodynamics of peptide formation, respectively. Given these developments, the simulation Three major developments have positioned the simulation community now has the tools to verify and validate simulation community to make significant advances toward under- protocols and models (forcefields). standing the mechanism of folding of peptides and small proteins. Firstly, the design of small peptides that mimic Addresses proteins in complexity, but are sufficiently small to allow Theoretical Biology and Biophysics Group, Theoretical Division, T10 MS detailed simulation studies [1–4]. Secondly, the develop- K710, Los Alamos National Laboratory, Los Alamos, NM 87545, USA à ment of fast (nanosecond) time-resolved spectroscopy e-mail: [email protected] methods to study peptide folding dynamics on the same timescale as computer simulations [5–8,9,10,11,12]. -

Beta-Hairpin Protein Epitope Mimetic Inhibitors of the P53-HDM2 Protein-Protein Interaction
Zurich Open Repository and Archive University of Zurich Main Library Strickhofstrasse 39 CH-8057 Zurich www.zora.uzh.ch Year: 2012 -Hairpin protein epitope mimetic technology in drug discovery Obrecht, Daniel ; Chevalier, Eric ; Moehle, Kerstin ; Robinson, John A Abstract: Epitopes involved in protein–protein and protein–nucleic acid interactions provide ideal starting points for rational structure-based inhibitor design. The process of design and optimization of epitope mimetics is now emerging as an innovative new approach in drug discovery. Although often derided as unsuitable for drug development, we provide examples to show how peptidomimetics can provide a new generation of drug candidates to tackle some of the most challenging targets in pharmaceutical research, and address some of the most pressing current threats to human health. DOI: https://doi.org/10.1016/j.ddtec.2011.07.006 Posted at the Zurich Open Repository and Archive, University of Zurich ZORA URL: https://doi.org/10.5167/uzh-69031 Journal Article Accepted Version Originally published at: Obrecht, Daniel; Chevalier, Eric; Moehle, Kerstin; Robinson, John A (2012). -Hairpin protein epitope mimetic technology in drug discovery. Drug Discovery Today: Technologies, 9(1):e63-e69. DOI: https://doi.org/10.1016/j.ddtec.2011.07.006 1 ß-Hairpin Protein Epitope Mimetic Technology in Drug Discovery Daniel Obrecht1 Eric Chevalier1 Kerstin Moehle2 and John A. Robinson2 1 Polyphor AG, Hegenheimermattweg 125, 4123-Allschwil, Switzerland (www.polyphor.com) 2 Chemistry Department, University of Zurich, Winterthurerstrasse 190, 8057-Zurich, Switzerland. (www.oci.uzh.ch) Corresponding author: Prof. J. A. Robinson Chemistry Department University of Zurich Winterthurerstrasse 190 8057 Zurich, Switzerland Phone: +41-44-6354242 Fax: +41-44-6356812 e-mail: [email protected] AND e-mail: [email protected] 2 Abstract Epitopes involved in protein-protein and protein-nucleic acid interactions provide ideal starting points for rational structure-based inhibitor design. -

Modeling the Mechanism of CLN025 Beta-Hairpin Formation
bioRxiv preprint doi: https://doi.org/10.1101/145185; this version posted June 19, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Modeling the Mechanism of CLN025 Beta-Hairpin Formation Keri A. McKiernana,1, Brooke E. Husica,1, and Vijay S. Pandea aDepartment of Chemistry, Stanford University, Stanford CA 94305, USA This manuscript was compiled on June 19, 2017 Beta-hairpins are a substructure found in proteins that can lend in- break in the folding mechanism of this protein at 308 K (8). sight into more complex systems. Furthermore, the folding of beta- Above this temperature, folding can no longer be described hairpins is a valuable test case for benchmarking experimental and using a two-state model. Because the experimental description theoretical methods. Here, we simulate the folding of CLN025, a of this system is both detailed and nontrivial at high tempera- miniprotein with a beta-hairpin structure, at its experimental melt- ture, we have benchmarked a set of popular protein force fields ing temperature using a range of state-of-the-art protein force fields. in their ability to describe the conformational dynamics of We construct Markov state models in order to examine the thermo- CLN025 at its experimental melting temperature of 340 K (7). dynamics, kinetics, mechanism, and rate-determining step of folding. The folding of CLN025 is of additional interest due to Mechanistically, we find the folding process is rate-limited by the for- its beta-hairpin structure. Neither experimentalists nor the- mation of the turn region hydrogen bonds, which occurs following orists have reached a consensus on the mechanism or rate- the downhill hydrophobic collapse of the extended denatured pro- determining step of beta hairpin folding (9–11). -

CHAPTER 6 Levels of Protein Structure Forces Contributing To
BCH 4053 Spring 2001 Chapter 6 Lecture Notes Slide 1 CHAPTER 6 Proteins: Secondary, Tertiary, and Quaternary Structure Slide 2 Levels of Protein Structure • Primary (sequence) • Secondary (ordered structure along peptide bond) • Tertiary (3 -dimensional overall) • Quaternary (subunit relationships) Slide 3 Forces Contributing to Overall Structure • Strong (peptide bond, disulfide bond) • Weak • Hydrophobic (40 kJ/mol) • Ionic bonds (~20 kJ/mol) • Figure 6.1 • Hydrogen bonds (~12-30 kJ/mol) • Dispersion (van der Waals) (0.4-4 kJ/mol) Chapter 6, page 1 Slide 4 Effect of Sequence on Structure • Sufficient information for folding into correct 3-dimensional structure is in the sequence (primary structure) of the protein • Experiments of Anfinsen and White on Ribonuclease • However—the “folding problem” is one of the major unsolved problems of biochemistry and structural biology Slide 5 Secondary Structure • Folding probably begins with nucleation sites along the peptide chain assuming certain stable secondary structures. • Planarity of the peptide bond restricts the number of conformations of the peptide chain. Rotation is only possible about the • C(alpha)-N bond (the F (phi) angle) • C(alpha)-C bond (the Y (psi) angle) • See Figure 6.2 Slide 6 Steric Constraints on F and YAngles • Examine the effects of rotation about the F and Y angles using Kinemage • Download Kinemage • Download Peptide file • Note that some angles are precluded by orbital overlap: • Figure 6.3 Chapter 6, page 2 Slide 7 Ramachandran Map • Plot of F versus Y angle for a peptide bond is called a Ramachandran Map • Ordered secondary structures have repeats of the F and Y angles along the chain.