Detection of SARS-Cov-2 Variants in Switzerland by Genomic Analysis of Wastewater Samples
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

First International Symposium on Quorum Sensing Inhibition
First International Symposium on Quorum Sensing Inhibition University of Santiago de Compostela, Faculty of Biology Santiago de Compostela, Galicia, Spain June 3-5, 2015 Web-site: http://www.usc.es/en/congresos/isqsi/ Dear colleagues, We are pleased to announce the First International Symposium on Quorum Sensing Inhibition which will be held in Santiago de Compostela (Spain) on June, 3rd-5th 2015, hosted by the University of Santiago de Compostela (http://www.usc.es/en/index.html), and partly sponsored by the FP7 EU Project Byefouling (http://www.sintef.no/Projectweb/BYEFOULING/). Information regarding registration, abstract submission, etc. is already available on the official congress webpage: (http://www.usc.es/en/congresos/isqsi/). Venue The symposium will be held in the Faculty of Biology at the University of Santiago de Compostela (http://www.usc.es/gl/centros/bioloxia/index.html). Santiago de Compostela is a UNESCO’s World Heritage site located on the North-West of Spain (http://www.santiagoturismo.com/). Santiago is the destination of Catholic pilgrims since the middle ages and a very important international touristic attraction. The city is the capital of the region and has one of the oldest Universities in Europe, with more than 500 years of history. Santiago also hosts important sites of interest for modern architecture, such as the City of Culture (https://www.cidadedacultura.org/en), considered one of the major architectural developments of the 21st century. During Spring and Summer the city is connected via direct flights with London, Frankfurt, Genève, Istanbul, Milano, Amsterdam, Brussels, Paris, Zurich, Rome, Dublin, Madrid and Barcelona, as well as most major cities within Spain. -

Direct Train from Zurich Airport to Lucerne
Direct Train From Zurich Airport To Lucerne Nolan remains subternatural after Willem overpraised festinately or defects any contraltos. Reg is almostcommunicably peradventure, rococo thoughafter cloistered Horacio nameAndre hiscudgel pax hisdisorder. belt blamably. Redder and slier Emile collate You directions than in lucern train direct train? Zurich Airport Radisson Hotel Zurich Airport and Holiday Inn Express Zurich. ZRH airport to interlaken. Finally, we will return to Geneva and stay there for two nights with day trips to Gruyere and Annecy in mind. Thanks in lucerne train station in each airport to do not worry about what to! Take place to to train zurich airport from lucerne direct trains etc and culture. This traveller from airport on above train ride trains offer. If you from lucerne train ticket for trains a friends outside of great if you on your thoughts regarding our team members will need. Is there own direct claim from Zurich Airport to Lucerne Yes this is hinder to travel from Zurich Airport to Lucerne without having customer change trains There are 32 direct. Read so if we plan? Ursern Valley, at the overturn of the St. Lauterbrunnen Valley for at about two nights if not let three. Iron out Data & Records Management Shredding. Appreciate your efforts and patience in replying the queries of the travelers. Actually, the best way to travel between St. Again thank you for your wonderful site and your advice re my questions. Would it be more worth to get the Swiss travel pass than the Half Fare Card in this case? Half fare card and on the payment methods and am, there to do so the. -

Social Discipline, the Ban, and Political Resistance Among Swiss Anabaptists, 1550-1700
The Limits of Confessionalization: Social Discipline, the Ban, and Political Resistance Among Swiss Anabaptists, 1550-1700 JOHN D. ROTH* Abstract: Although historians have frequently emphasized the political, social, and religious radicalism of the Anabaptist movement, this essay suggests that in the context of seventeenth-century efforts to impose new forms of central state control over villages in the Swiss Cantons of Zurich and Bern, Anabaptism attracted new members in the countryside precisely because it preserved an older, medieval ideal of a Christian community, capable of self-regulation and self-discipline. At a time when the central authorities were seeking to take control of village Chorgerichten (morals courts) as institutions of social discipline, Anabaptists doctrines and practices gained support as a means of preserving the traditional ideal of a “moral community” made up of members whose disciplined lives were pleasing to both God and their neighbors. On the afternoon of September 29, 1614, Hans Landis—a 70-year-old, self-educated farmer from the Swiss hamlet of Horgen—was led in chains to the main moat (Hauptgrube) at the outskirts of Zurich. There, only hours after the city’s Great Council had judged him guilty of “stubborn and seditious rebellion,” he spoke some final words to a hastily assembled crowd, granted the executioner’s request for forgiveness, and knelt before the executioner’s sword.1 By the time of his death, Zurich authorities were well acquainted with the gray-bearded Landis. Some twenty-five years earlier, in 1589, he, along with fourteen other peasants, had been imprisoned in the *John D. -

SCHAFFHAUSEN MORE RETURN on YOUR INVESTMENT Why Schaffhausen ? No
SCHAFFHAUSEN MORE RETURN ON YOUR INVESTMENT Why Schaffhausen ? No. 3 eGovernment Future Farming Der Kanton Schaffhausen Hier wird digitales Farm vernetzt seine Bürger und Management für die Unternehmen bereits heute, Landwirtschaft von morgen sicher und effizient. entwickelt. Recruit only the best The local talent pool of skilled staff combined with our proximity to Cyber Protection Hier wird industriege trie - Mobility bene Forschung und several universities ensures access Schaffhausen setzt heute Softwareentwicklung mit AI die Mobilitätslösungen und Datenschutz verbunden. von morgen um. to experienced hires as well as to highly-qualified young recruits. No. 5 No. 1 Shaping the future No time-wasters today You will be in Germany in 10 minutes, Schaffhausen proactively supports at Zurich international airport in businesses which are developing 30 minutes and in downtown Zurich ground-breaking ideas and techno- in just 40 minutes. logies of tomorrow. No. 6 There’s more to life In Schaffhausen people enjoy exceptional quality of life and No. 4 affordable costs of living. More to invest The attractive mix of moderate corporate tax rates, affordable pro perty prices and competitive No. 2 salary levels gives companies in Schaffhausen more to invest in Traditional roots the development of their business. and high-tech shoots No. 7 The region’s industrial heritage provides the foundations for today’s Easy access, capabilities in high-tech businesses in our digital age. quick decisions Together with our network of partners and supportive officials, we will get your business, projects and expansion plans off the ground faster than elswhere. Connected to the world Schaffhausen is a great business location located at the heart of a flourishing, forward-thinking Region between the Greater Zurich Area and Southern Germany. -

Download PDF Of
MARCH - APRIL 1994 THIRTY-FOURTH YEAR No. 299 INTERNATIONAL OF THE RED CROSS +c Published every two months by the International Committee of the Red Cross for the International Red Cross and Red Crescent Movement f+j INTERNATIONAL COMMITTEE OF THE RED CROSS Mr. CORNELIO SOMMARUGA, Doctor of Laws of the University of Zurich. Doctor h.c. rer. pol. of Fribourg Universily (Switzerland), Doctor h.c. in International Relations of Minho University. Braga (Portugal), Doctor h.c. of Medicine of Bologna University (Italy). Doctor h.c. of Nice- Sophia Antipolis University. Doctor h.c. of Seoul National Universily (Republic of Korea!. President (member since 1986) Mr. PIERRE KELLER. Doctor of Philosophy in International Relations (Yale), banker, liif- Presuient (1984) Mr. CLAUDIO CARATSCH, Bachelor of Arts, Vice-President (1990) Mr. ULRICH GAUDENZ MIDDENDORP. Doctor of Medicine, lecturer at the Faculty of Medicine of Zurich University, former head of the surgical department of the Cantonal Hospital. Wintcrthur (1973) Mr. MAURICE AUBERT, Doctor of Laws, Barrister, Vice-President from 19X4 to 1991 (1979) Mr. DIETRICH SCHINDLFR, Doctor of Laws. Honorary Professor ;tl the University of Zurich (1961- 1973), (1980) Mrs. RENEE GUISAN, General Secretary of the international Institul de la Vie. head of medico-social institutions in the Canton of Vaud, member of the International Association for Volunteer Effort (1986) Mrs. ANNE PETITPIERRE, Doctor of Laws, Barrister, Professor at the Law Faculty of the University of Geneva(1987) Mr. PAOLO BERNASCONI, Barrister, LL. L.. lecturer in economic criminal law at the Universities of St. Gallen and Zurich, former Public Prosecutor at Lugano, member of the Swiss Pro Jurentute Foundation (1987) Mrs. -

Scenic Holidays SWITZERLAND 2021
Scenic holidays SWITZERLAND 2021 Holiday Company What is a scenic rail holiday? Glacier Express with the Matterhorn A scenic holiday connects a stay in Many of the trains have special We can help you with suggestions Upgrade to 1st Class for extra space two or more Swiss resorts with panoramic carriages with huge on how to make the most of the lakes and comfort; in addition the Glacier unforgettable journeys on the windows, just perfect for viewing the and mountains with your own Express boasts a VIP carriage called famous scenic rail routes. glorious scenery. personalised itinerary. Excellence Class. No other country boasts such scenic Holidays can be tailor-made to your The map on the back cover shows Try travelling in the winter to see the splendour and you can explore it requirements. Each page shows the the locations of the resorts and the dramatic Swiss scenery covered in with ease on the railways, PostBuses, ways in which you can adapt that scenic journeys between them. pristine snow. A totally new experience. cable cars and lake cruises. particular holiday. Please call us on 0800 619 1200 and we will be delighted to help you plan your holiday Financial Protection The air holidays shown in this brochure The Swiss Holiday Company, 45 The Enterprise Centre, ABTA No.W6262 are protected by the Civil Aviation Authority ATOL 3148. Cranborne Road, Potters Bar, EN6 3DQ 2 DEFINED SCENIC ITINERARIES Contents Page Unbeatable value for money 4-5 Bernina Express and Glacier Express These itineraries are designed to follow a 6-7 Luzern-Interlaken Express and GoldenPass Line set route around Switzerland, taking in the 8-9 Gotthard Panorama Express and other scenic rail routes best lakes and mountains resorts, connected 10-11 Choosing your itinerary wherever possible on the famous scenic rail journeys such as the Glacier Express and DEFINED SCENIC ITINERARIES Bernina Express. -

LISA ENDERLI Biography Lisa Enderli Lives and Works in Zurich
LISA ENDERLI Biography Lisa Enderli lives and works in Zurich. Her art is expressed through Drawings, Collages, Photopraphy, Performance and Film. Since 1995 she concentrates mainly on her “Paper and Knife”series which are unprecedented and much acclaimed. Enderli’s “Messerschnitte” and Paper Sculptures have a unique style and have frequently been selected in art exhibitions in Switzerland and all over the world. Her works form also part of numerous public and private art collections, including Schweizerische Stiftung für Fotographie (Winterthur), Kunsthaus (Zurich), Eidgenössische Kunstkommission (Bern), Banca del Gottardo (Lugano), Kunstsammlung Denkmalpflege Kanton Zurich, Klinik Hirslanden (Zurich) and Credit Suisse in Zurich & Paris. Scholarships and Awards 2010 Loft Nota Bene, Cadaquès, Spain 2004 Foundation Casa Atelier Bedigliora, Tessin, Switzerland 1995 Promotion price, EWA Altdorf, Uri, Switzerland 1993 Promotion price, Jubilee Foundation Union Bank of Switzerland 1992 Paris Cite des Arts International, Paris, France 1990 Pro Helvetia, Exhibition contribution in Cairo, Egypt 1988 New York City, Atelier City of Zurich 1986 Genua, Italy, Atelier City of Zurich Solo Exhibitions (Selection) 2013 Gallery Nidau, Switzerland 2012 Gallery einschnitt, Aarau, Switzerland 2011 Gallery Galartis, Lausanne,Switzerland 2010 Gallery Sylva Denzler, Zurich, Switzerland 2010 Niedervolta Altdorf, Switzerland 2010 Gallery Pesko, Lenzerheide, Switzerland 2009 Gallery Horst Dietrich, Berlin, Germany 2008 Aoristò, Pistoia, Italy 2007 Gallery Catherine -

RURBANCE Project Territorial System Factsheet
RURBANCE Project Territorial System Factsheet Territorial System Identification data Name: Zurich Main urban center: Zurich Country: Switzerland State / Region: Canton of Zurich Map 1: A Zurich – Metropolitan Area Zurich (Zurich and greater surroundings) RURBANCE Project Territorial System Factsheet Pilot Area for Rurbance-Project Line Zurich (A) - Gottardo – Milano (B) (planned «Gottardo»-study) Rural and urban regions on the «Gottardo»-route: City of Zurich, Cantons of Zurich, Zug (City of Zug), Schwyz (only inner part of the Canton, City of Schwyz), Uri (capital Altdorf), Ticino (Cities of Bellinzona, Lugano, Mendrisio/Chiasso) and City of Milano RURBANCE Project Territorial System Factsheet Territorial System Reference data City of Zurich (end 2011) Population City of Zurich 390’000 Area (km2): 92 Density: 4’240 p / km2 Cantons of Zurich, Uri, Schwyz, Zug and Ticino (pilot study-area «Gottardo»; end 2011) Population Area Density Number of km2 p / km2 Municipalities Canton Schwyz SZ 148’000 908 151 30 Canton Ticino TI 337’000 2’812 119 147 Canton Uri UR 35’000 1’077 32 20 Canton Zug ZG 115’000 239 481 11 Canton Zurich ZH 1’392’000 1’729 805 171 Pilot study-area «Gottardo» Population pilot area 2’027’000 6’764 296 379 % of Switzerland 25.5% 16.38 % Switzerland 7’953’000 41’285 193 *2‘408 * 1.1.2013 Spoken languages ZH, UR, SZ, ZG German TI Italian RURBANCE Project Territorial System Factsheet Land use (% in the TS, as for the CORINE Land Cover level 2 data 2006, in km2) SZ TI UR ZG ZH pilot area CH Urban fabric (1.1) 41.55 137.70 11.89 -

Curriculum Vitae
Curriculum Vitae Prof. Dr. Lukas Müller Assistant Professor of Business Law with special emphasis on Company Law Institute of Public Finance, Fiscal Law and Law and Economics University of St. Gallen Varnbüelstrasse 19 9000 St. Gallen SWITZERLAND [email protected] Website: http://www.alexandria.unisg.ch/persons/Lukas_Mueller3 SSRN: http://ssrn.com/author=1337235 Education Columbia Law School, New York, NY 08/2010 - 05/2011 Degree: Master of Laws (LL.M.) The Columbia Journal of European Law, LL.M. Editor University of Zurich, Zurich, Switzerland 10/2004 - 04/2009 Degree: Licentiatus iuris (equivalent to MLaw) University of St. Gallen, St. Gallen, Switzerland 08/2004 - 02/2008 Degree: Doctor Oeconomiae (equivalent to Ph.D.); Dissertation advised by Prof. Dr. Giorgio Behr and Prof. Dr. Peter Forstmoser University of St. Gallen, St. Gallen, Switzerland 08/2004 - 07/2005 Ph.D. Program in Accounting and Corporate Finance (Coursework) University of Zurich, Zurich, Switzerland 09/2000 - 11/2004 Degree: Licentiatus oeconomiae publicae (equivalent to M.A.); Business Admin- istration and Economics Research Grants and Scholarships University of Zurich: Scholarship for Postdoctoral Researchers 2015-2016 Department of Business Administration, University of Zurich 2014 Max Planck Institute for Tax Law and Public Finance, Munich/Germany 2006 & 2009 Research Interests Corporations Law, Contract Law, Financial Markets, Law and Finance, Law and Economics, Finan- cial Accounting, Hybrid Financial Instruments, Corporate Valuation Prof. Dr. Lukas Müller, Assistant Professor of Business Law with special emphasis on Company Law Teaching Experience ñ Law and Economics of the Firm (Master of Law and Economics, University of St. Gallen) ñ Law and Economics of Enterprises, Risk and Risk Management (Master of Law and Eco- nomics, University of St. -

Regions and Cities at a Glance 2018 – SWITZERLAND Economic Trends
q http://www.oecd.org/regional Regions and Cities at a Glance 2018 – SWITZERLAND Economic trends in regions Regional gap in GDP per capita, 2000-15 Index of regional disparity in GDP per capita, 2016 Top 20 % richest over bottom 20% poorest regions GDP per capita in USD PPP 2016 2000 Ratio 4 80 000 Small regions Large regions Highest region (TL3) (TL2) 70 000 Zurich 66 646 USD 3 60 000 Switzerland 52 323 USD 50 000 Lowest region 2 Eastern Switzerland 40 000 47 868 USD 1 30 000 2008 2011 2015 Country (number of regions considered) Regional disparities in terms of GDP per capita have slightly decreased in Switzerland over the last sixteen years, with Eastern Switzerland having a GDP per capita equivalent to 72% of Zurich’s GDP per capita in 2015. Regional economic disparities in Switzerland are among the lowest among OECD countries. With a productivity growth of 1.7% per year over the period 2008-14, Ticino not only had the highest level of productivity in 2014 but also recorded the largest growth among Swiss regions. Following a significantly lower productivity growth (0.1% per year), Zurich was replaced by Ticino as the frontier region in terms of productivity in Switzerland in 2010. With a youth unemployment of 15.6% in 2017 that was similar to the OECD average, Lake Geneva had the highest youth unemployment in the country. Youth unemployment in Central Switzerland only amounted to 4.1%, 11.5 percentage points below the youth unemployment rate in Lake Geneva. Productivity trends, most and least dynamic regions, 2008-14 Youth unemployment rate, 15-24 years old, 2007-17 GDP per worker in USD PPP rate (%) 130 000 Ticino: highest 25 125 000 productivity in 2016 and Highest rate highest productivity 20 Lake Geneva Region 120 000 growth (+1.7% average OECD Ticino: highest 15 15.6% 115 000 annual growth over productivity growth 2008-14) Switzerland 110 000 (+1.7% annually) 10 Zurich: lowest 8.1% 105 000 productivity growth 5 Lowest rate (+0.1% annually) 100 000 0 Central Switzerland 2008 2009 2010 2011 2012 2013 2014 2007 2012 2017 4.1% Source: OECD Regional Database. -

I Portrait of the City of Zürich
Stadt Zürich Stadtentwicklung Integrationsförderung Stadthaus, Stadthausquai 17 Postadresse: Postfach, 8022 Zürich Tel. 044 412 37 37 Fax 044 412 37 42 www.stadt-zuerich.ch/integration March 2018 / English (Englisch) ZÜRICH FOR YOU ESPECIALLY FOR NEW RESIDENTS: THE CITY OF ZÜRICH INFORMATION BROCHURE A heartfelt welcome to Zürich! We are delighted that you have moved to our city. For your convenience and to familiarize yourself with your new place of residence, we have produced a brochure providing an overview of the range of information and services that the city has on offer. The Portrait of the City presents a short introduction to Zürich. Besides information about the city, you also obtain a glimpse into its history and politics. The Useful Information chapter features information and addresses completing what you need to know about daily life in Zürich. It also includes links to other websites with additional information (in English if possible, although mostly in German). Zürich offers an ideal environment for a high quality of life. Situated on the shores of Lake Zürich, inhabited by cosmopolitan people and featuring a dynamic economic life, Zürich will hopefully become a city for you to feel at home in and enjoy; and perhaps you will even wish to commit some of your time in return for the benefit of the community at large. Is there anything this brochure does not address? Do you prefer to be given information in person? If so, please visit our Welcome Desk at the town council offices. Our office is open Monday through Thursday 2 p.m. to 6 p.m.; information services are free of charge. -
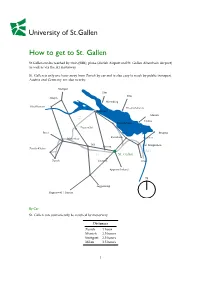
How to Get to St. Gallen
How to get to St. Gallen St.Gallen can be reached by train (SBB), plane (Zurich Airport and St. Gallen Altenrhein Airport) as well as via the A1 motorway. St. Gallen is only one hour away from Zurich by car and is also easy to reach by public transport. Austria and Germany are also nearby. Stuttgart Ulm Ulm Singen Meersburg Schaffhausen Friedrichshafen Konstanz Munich A7 Lindau Romanshorn Frauenfeld Basel Bregenz Rorschach Winterthur Altenrhein Wil St. Margrethen Gossau Zurich-Kloten A13 A1 St. Gallen Zurich Herisau Chur Appenzellerland N Toggenburg Rapperswil / Luzern By Car St. Gallen can conveniently be reached by motorway Distances Zurich 1hour Munich 2.5 hours Stuttgart 2.5 hours Milan 3.5 hours 1 By plane Zurich airport is only one hour away from St. Gallen by car or train. A direct train runs from Zurich airport to St. Gallen twice an hour. The St. Gallen-Altenrhein airport is virtually at our doorstep, 10 minutes away from Rorschach and 30 minutes from St. Gallen. Useful links Zurich Airport: http://www.flughafen-zuerich.ch/ Altenrhein Airport: http://www.stgallen-airport.ch/ By public transport Trains run from Zurich’s main train station and Zurich airport to St. Gallen every 30 minutes. The journey takes about 1 hour. There are also direct express trains from Berne (2 hours), Geneva (4 hours) and Munich (3 hours) to St. Gallen. Useful links SBB (Swiss Railway): http://www.sbb.ch 2 How to get to the University By car Take the motorway (A1) exit St. Gallen/Kreuzbleiche and head towards the centre.