SMART-DISSERTATION-2016.Pdf (2.356Mb)
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Herpetofauna Diversity from Khamgaon, District Buldhana (M.S.) Central India
Int. J. of Life Sciences, 2016, Vol. 4 (3): 412-418 ISSN: 2320-7817| eISSN: 2320-964X RESEARCH ARTICLE Herpetofauna Diversity from Khamgaon, district Buldhana (M.S.) Central India Bawaskar Prakas S1 and Bawaskar Kiran S2 1P.G. Department of Zoology G.S. College, Khamgaon-444303, Dist-Buldana (M.S.) India. 2Department of Chemistry Narasamma Hirayya College, Kiran nagar , Amravati 444601. *Corresponding author Email: [email protected] | [email protected] Manuscript details: ABSTRACT Received: 21.09.2016 Preliminary checklist of Herpetofauna diversity from Khamgaon taluk, Accepted: 05.10.2016 district Buldhana (M.S.) Central India at co-ordinate Latitude 20.6833, Published : 03.11.2016 Longitude 76.5666. In present there is no report on Herpetofauna diversity from Khamgaon taluk, so the present study has been carried Editor: Dr. Arvind Chavhan out during 2010-2015 in an alternatively days and nights herping. The study area covers such as Marshes, grass lands, rocky area, farm lands, Cite this article as: scrub lands, forest, hilly area, villages and town. It was observed that 13 Bawaskar Prakas S and Bawaskar species found to be an abundant, 14 species were common, 12 species Kiran S (2016) Herpetofauna were uncommon, 9 species were occasional and 10 species were found Diversity from Khamgaon, district Buldhana (M.S.) Central India, to a rare. A general trend increased Herpetofauna population was International J. of Life Sciences, 4 (3): observed in monsoon while comparatively less population of 412-418. Herpetofauna observed during a winter to early monsoon. Acknowledgements: Key words: Diversity, Herping, Herpetofauna, Khamgaon. Authors are especially thankful to Snake friends and Wildlifers community including Mr. -

A Molecular Phylogeny of the Lamprophiidae Fitzinger (Serpentes, Caenophidia)
Zootaxa 1945: 51–66 (2008) ISSN 1175-5326 (print edition) www.mapress.com/zootaxa/ ZOOTAXA Copyright © 2008 · Magnolia Press ISSN 1175-5334 (online edition) Dissecting the major African snake radiation: a molecular phylogeny of the Lamprophiidae Fitzinger (Serpentes, Caenophidia) NICOLAS VIDAL1,10, WILLIAM R. BRANCH2, OLIVIER S.G. PAUWELS3,4, S. BLAIR HEDGES5, DONALD G. BROADLEY6, MICHAEL WINK7, CORINNE CRUAUD8, ULRICH JOGER9 & ZOLTÁN TAMÁS NAGY3 1UMR 7138, Systématique, Evolution, Adaptation, Département Systématique et Evolution, C. P. 26, Muséum National d’Histoire Naturelle, 43 Rue Cuvier, Paris 75005, France. E-mail: [email protected] 2Bayworld, P.O. Box 13147, Humewood 6013, South Africa. E-mail: [email protected] 3 Royal Belgian Institute of Natural Sciences, Rue Vautier 29, B-1000 Brussels, Belgium. E-mail: [email protected], [email protected] 4Smithsonian Institution, Center for Conservation Education and Sustainability, B.P. 48, Gamba, Gabon. 5Department of Biology, 208 Mueller Laboratory, Pennsylvania State University, University Park, PA 16802-5301 USA. E-mail: [email protected] 6Biodiversity Foundation for Africa, P.O. Box FM 730, Bulawayo, Zimbabwe. E-mail: [email protected] 7 Institute of Pharmacy and Molecular Biotechnology, University of Heidelberg, INF 364, D-69120 Heidelberg, Germany. E-mail: [email protected] 8Centre national de séquençage, Genoscope, 2 rue Gaston-Crémieux, CP5706, 91057 Evry cedex, France. E-mail: www.genoscope.fr 9Staatliches Naturhistorisches Museum, Pockelsstr. 10, 38106 Braunschweig, Germany. E-mail: [email protected] 10Corresponding author Abstract The Elapoidea includes the Elapidae and a large (~60 genera, 280 sp.) and mostly African (including Madagascar) radia- tion termed Lamprophiidae by Vidal et al. -

Snakes of South-East Asia Including Myanmar, Thailand, Malaysia, Singapore, Sumatra, Borneo, Java and Bali
A Naturalist’s Guide to the SNAKES OF SOUTH-EAST ASIA including Myanmar, Thailand, Malaysia, Singapore, Sumatra, Borneo, Java and Bali Indraneil Das First published in the United Kingdom in 2012 by Beaufoy Books n n 11 Blenheim Court, 316 Woodstock Road, Oxford OX2 7NS, England Contents www.johnbeaufoy.com 10 9 8 7 6 5 4 3 2 1 Introduction 4 Copyright © 2012 John Beaufoy Publishing Limited Copyright in text © Indraneil Das Snake Topography 4 Copyright in photographs © [to come] Dealing with Snake Bites 6 All rights reserved. No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, without the prior written permission of the publishers. About this Book 7 ISBN [to come] Glossary 8 Edited, designed and typeset by D & N Publishing, Baydon, Wiltshire, UK Printed and bound [to come] Species Accounts and Photographs 11 Checklist of South-East Asian Snakes 141 Dedication Nothing would have happened without the support of the folks at home: my wife, Genevieve V.A. Gee, and son, Rahul Das. To them, I dedicate this book. Further Reading 154 Acknowledgements 155 Index 157 Edited and designed by D & N Publishing, Baydon, Wiltshire, UK Printed and bound in Malaysia by Times Offset (M) Sdn. Bhd. n Introduction n n Snake Topography n INTRODUCTION Snakes form one of the major components of vertebrate fauna of South-East Asia. They feature prominently in folklore, mythology and other belief systems of the indigenous people of the region, and are of ecological and conservation value, some species supporting significant (albeit often illegal) economic activities (primarily, the snake-skin trade, but also sale of meat and other body parts that purportedly have medicinal properties). -

Nyika and Vwaza Reptiles & Amphibians Checklist
LIST OF REPTILES AND AMPHIBIANS OF NYIKA NATIONAL PARK AND VWAZA MARSH WILDLIFE RESERVE This checklist of all reptile and amphibian species recorded from the Nyika National Park and immediate surrounds (both in Malawi and Zambia) and from the Vwaza Marsh Wildlife Reserve was compiled by Dr Donald Broadley of the Natural History Museum of Zimbabwe in Bulawayo, Zimbabwe, in November 2013. It is arranged in zoological order by scientific name; common names are given in brackets. The notes indicate where are the records are from. Endemic species (that is species only known from this area) are indicated by an E before the scientific name. Further details of names and the sources of the records are available on request from the Nyika Vwaza Trust Secretariat. REPTILES TORTOISES & TERRAPINS Family Pelomedusidae Pelusios rhodesianus (Variable Hinged Terrapin) Vwaza LIZARDS Family Agamidae Acanthocercus branchi (Branch's Tree Agama) Nyika Agama kirkii kirkii (Kirk's Rock Agama) Vwaza Agama armata (Eastern Spiny Agama) Nyika Family Chamaeleonidae Rhampholeon nchisiensis (Nchisi Pygmy Chameleon) Nyika Chamaeleo dilepis (Common Flap-necked Chameleon) Nyika(Nchenachena), Vwaza Trioceros goetzei nyikae (Nyika Whistling Chameleon) Nyika(Nchenachena) Trioceros incornutus (Ukinga Hornless Chameleon) Nyika Family Gekkonidae Lygodactylus angularis (Angle-throated Dwarf Gecko) Nyika Lygodactylus capensis (Cape Dwarf Gecko) Nyika(Nchenachena), Vwaza Hemidactylus mabouia (Tropical House Gecko) Nyika Family Scincidae Trachylepis varia (Variable Skink) Nyika, -

Reptile Rap Newsletter of the South Asian Reptile Network ISSN 2230-7079 No.15 | January 2013 Date of Publication: 22 January 2013 1
Reptile Rap Newsletter of the South Asian Reptile Network No.15 | January 2013 ISSN 2230-7079 Date of publication: 22 January 2013 1. Crocodile, 1. 2. Crocodile, Caiman, 3. Gharial, 4.Common Chameleon, 5. Chameleon, 9. Chameleon, Flap-necked 8. Chameleon Flying 7. Gecko, Dragon, Ptychozoon Chamaeleo sp. Fischer’s 10 dilepsis, 6. &11. Jackson’s Frill-necked 21. Stump-tailed Skink, 20. Gila Monster, Lizard, Green Iguana, 19. European Iguana, 18. Rhinoceros Antillean Basilisk, Iguana, 17. Lesser 16. Green 15. Common Lizard, 14. Horned Devil, Thorny 13. 12. Uromastyx, Lizard, 34. Eastern Tortoise, 33. 32. Rattlesnake Indian Star cerastes, 22. 31. Boa,Cerastes 23. Python, 25. 24. 30. viper, Ahaetulla Grass Rhinoceros nasuta Snake, 29. 26. 27. Asp, Indian Naja Snake, 28. Cobra, haje, Grater African 46. Ceratophrys, Bombina,45. 44. Toad, 43. Bullfrog, 42. Frog, Common 41. Turtle, Sea Loggerhead 40. Trionychidae, 39. mata Mata 38. Turtle, Snake-necked Argentine 37. Emydidae, 36. Tortoise, Galapagos 35. Turtle, Box 48. Marbled Newt Newt, Crested 47. Great Salamander, Fire Reptiles, illustration by Adolphe Millot. Source: Nouveau Larousse Illustré, edited by Claude Augé, published in Paris by Librarie Larousse 1897-1904, this illustration from vol. 7 p. 263 7 p. vol. from 1897-1904, this illustration Larousse Librarie by published in Paris Augé, Claude by edited Illustré, Larousse Nouveau Source: Millot. Adolphe by illustration Reptiles, www.zoosprint.org/Newsletters/ReptileRap.htm OPEN ACCESS | FREE DOWNLOAD REPTILE RAP #15, January 2013 Contents A new record of the Cochin Forest Cane Turtle Vijayachelys silvatica (Henderson, 1912) from Shendurney Wildlife Sanctuary, Kerala, India Arun Kanagavel, 3–6pp New Record of Elliot’s Shieldtail (Gray, 1858) in Seshachalam Biosphere Reserve, Eastern Ghats, Andhra Pradesh, India M. -

Marine Reptiles Arne R
Virginia Commonwealth University VCU Scholars Compass Study of Biological Complexity Publications Center for the Study of Biological Complexity 2011 Marine Reptiles Arne R. Rasmessen The Royal Danish Academy of Fine Arts John D. Murphy Field Museum of Natural History Medy Ompi Sam Ratulangi University J. Whitfield iG bbons University of Georgia Peter Uetz Virginia Commonwealth University, [email protected] Follow this and additional works at: http://scholarscompass.vcu.edu/csbc_pubs Part of the Life Sciences Commons Copyright: © 2011 Rasmussen et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Downloaded from http://scholarscompass.vcu.edu/csbc_pubs/20 This Article is brought to you for free and open access by the Center for the Study of Biological Complexity at VCU Scholars Compass. It has been accepted for inclusion in Study of Biological Complexity Publications by an authorized administrator of VCU Scholars Compass. For more information, please contact [email protected]. Review Marine Reptiles Arne Redsted Rasmussen1, John C. Murphy2, Medy Ompi3, J. Whitfield Gibbons4, Peter Uetz5* 1 School of Conservation, The Royal Danish Academy of Fine Arts, Copenhagen, Denmark, 2 Division of Amphibians and Reptiles, Field Museum of Natural History, Chicago, Illinois, United States of America, 3 Marine Biology Laboratory, Faculty of Fisheries and Marine Sciences, Sam Ratulangi University, Manado, North Sulawesi, Indonesia, 4 Savannah River Ecology Lab, University of Georgia, Aiken, South Carolina, United States of America, 5 Center for the Study of Biological Complexity, Virginia Commonwealth University, Richmond, Virginia, United States of America Of the more than 12,000 species and subspecies of extant Caribbean, although some species occasionally travel as far north reptiles, about 100 have re-entered the ocean. -

Molecular Evolution of Three-Finger Toxins in the Long-Glanded Coral Snake Species Calliophis Bivirgatus
toxins Article Electric Blue: Molecular Evolution of Three-Finger Toxins in the Long-Glanded Coral Snake Species Calliophis bivirgatus Daniel Dashevsky 1,2 , Darin Rokyta 3 , Nathaniel Frank 4, Amanda Nouwens 5 and Bryan G. Fry 1,* 1 Venom Evolution Lab, School of Biological Sciences, University of Queensland, St Lucia, QLD 4072, Australia; [email protected] 2 Australian National Insect Collection, Commonwealth Science and Industry Research Organization, Canberra, ACT 2601, Australia 3 Department of Biological Sciences, Florida State University, Tallahassee, FL 24105, USA; [email protected] 4 MToxins Venom Lab, 717 Oregon Street, Oshkosh, WI 54902, USA; [email protected] 5 School of Chemistry and Molecular Biosciences, University of Queensland, St Lucia, QLD 4072, Australia; [email protected] * Correspondence: [email protected], Tel.: +61-7-336-58515 Abstract: The genus Calliophis is the most basal branch of the family Elapidae and several species in it have developed highly elongated venom glands. Recent research has shown that C. bivirgatus has evolved a seemingly unique toxin (calliotoxin) that produces spastic paralysis in their prey by acting on the voltage-gated sodium (NaV) channels. We assembled a transcriptome from C. bivirgatus to investigate the molecular characteristics of these toxins and the venom as a whole. We find strong confirmation that this genus produces the classic elapid eight-cysteine three-finger toxins, that δ-elapitoxins (toxins that resemble calliotoxin) are responsible for a substantial portion of the venom composition, and that these toxins form a distinct clade within a larger, more diverse clade of C. bivirgatus three-finger toxins. This broader clade of C. -

Ancestral Reconstruction of Diet and Fang Condition in the Lamprophiidae: Implications for the Evolution of Venom Systems in Snakes
Journal of Herpetology, Vol. 55, No. 1, 1–10, 2021 Copyright 2021 Society for the Study of Amphibians and Reptiles Ancestral Reconstruction of Diet and Fang Condition in the Lamprophiidae: Implications for the Evolution of Venom Systems in Snakes 1,2 1 1 HIRAL NAIK, MIMMIE M. KGADITSE, AND GRAHAM J. ALEXANDER 1School of Animal, Plant and Environmental Sciences, University of the Witwatersrand, Johannesburg. PO Wits, 2050, Gauteng, South Africa ABSTRACT.—The Colubroidea includes all venomous and some nonvenomous snakes, many of which have extraordinary dental morphology and functional capabilities. It has been proposed that the ancestral condition of the Colubroidea is venomous with tubular fangs. The venom system includes the production of venomous secretions by labial glands in the mouth and usually includes fangs for effective delivery of venom. Despite significant research on the evolution of the venom system in snakes, limited research exists on the driving forces for different fang and dental morphology at a broader phylogenetic scale. We assessed the patterns of fang and dental condition in the Lamprophiidae, a speciose family of advanced snakes within the Colubroidea, and we related fang and dental condition to diet. The Lamprophiidae is the only snake family that includes front-fanged, rear-fanged, and fangless species. We produced an ancestral reconstruction for the family and investigated the pattern of diet and fangs within the clade. We concluded that the ancestral lamprophiid was most likely rear-fanged and that the shift in dental morphology was associated with changes in diet. This pattern indicates that fang loss, and probably venom loss, has occurred multiple times within the Lamprophiidae. -

Venomous Snakes
Venomous Snakes - By Kedar Bhide Kedar Bhide is a snake expert from Mumbai. A postgraduate from Mumbai's Haffkine Institute, his work has resulted into first records of 2 snake species for India, Barta (Kaulback's Pit Viper) from Arunachal Pradesh and the Sind Awl-headed snake from Rajasthan. “ Moments after being bitten, the man feels a live fire germinating in the wound as if red hot tongs contorted his flesh; that which was mortified enlarges to monstrosity, and lividness invades him. The unfortunate victim witnesses his body becoming corpse piece by piece; a chill of death invades all his being, and soon bloody threads fall from his gums; and his eyes, without intending to, will also cry blood, until, beaten by suffering and anguish, he loses the sense of reality. If we then ask the unlucky man something, he may see us through blurred eyes, but we get no response; and perhaps a final sweat of red pearls or a mouthful of blackish blood warns of impending” (This is an introduction of a book written in 1931 by a Costa Rican Biologists and snakebite expert Clodomiro Picado.) INTRODUCTION Human fear of snakes is caused almost entirely by those species that can deliver a venomous bite. It is somewhat ironic that such a minority group, like venomous snakes has endangered the whole kingdom of snakes. Let us start by correcting a frequent misnomer. People often refer to poisonous snakes, and indeed by directory definition, this is not incorrect. But as a student of herpetology we should be more specific in our terminology. -

A New Colour Morph of Calliophis Bibroni (Squamata: Elapidae) and Evidence for Müllerian Mimicry in Tropical Indian Coralsnakes
Herpetology Notes, volume 10: 209-217 (2017) (published online on 25 April 2017) A new colour morph of Calliophis bibroni (Squamata: Elapidae) and evidence for Müllerian mimicry in Tropical Indian coralsnakes Dileep Kumar Raveendran1, V. Deepak2, Eric N. Smith3 and Utpal Smart3,* Abstract. Meristic and molecular data provide evidence for an exceptional multi-chromatic defensive strategy in an Indian coralsnake, Calliophis bibroni from the state of Kerala. We propose a mimicry hypothesis involving a combination of an ontogenetic colour shift at maturity, from initial Müllerian mimicry with a subtropical Indian coralsnake Sinomicrurus macclellandii, to one of two very different adult dorsal colours: 1) an aposematic pattern resembling that of the sympatric tropical Indian coralsnake Calliophis castoe or 2) a cryptic dark brown colouration. To this end, we succinctly juxtapose the rich body of work on mimicry in New World elapids to that of their Old World counterparts in an attempt to address the exciting yet unexplored prospect of investigating mimicry, crypsis and aposematism in Old World coralsnakes. Key Words. Aposematism, crypsis, Indian coralsnakes, genetic distance, meristics, mimicry Introduction Müllerian co-mimics benefit from sharing the same signal since this reduces the number of individuals that Animal colouration provides many functions; the most have to be sacrificed per prey species to educate local important among them is probably predator avoidance predators of a given aposematic colouration (Müller, and deterrence. Camouflage, or crypsis, blends animals 1879). Furthermore, possibly to balance prey-predator into their environment while deimatism surprises and dynamics, some mimics may replicate the colours confuses predators with the display of startling colour or patterns of dangerous models which lack bright, patterns. -

Amblyodipsas Polylepis (Bocage, 1873) Feeding on the Amphisbaenid Monopeltis Luandae Gans, 1976
Herpetology Notes, volume 14: 205-207 (2021) (published online on 26 January 2021) A snake with an appetite for the rare: Amblyodipsas polylepis (Bocage, 1873) feeding on the amphisbaenid Monopeltis luandae Gans, 1976 Werner Conradie1,* and Pedro Vaz Pinto2,3 Specimens in natural history museum collections catalogue number PEM R22034. The snake measured represent a unique snapshot of the time and place they 634 mm in snout–vent length (no tail length is provided were collected, while the analysis of stomach contents as the tail was truncated). Identification to the nominate often leads to unexpected results and new discoveries. For subspecies A. p. polylepis was based on a series of example, the Angolan lizard Ichnotropis microlepidota characteristics (fide Broadley, 1990), including enlarged Marx, 1956 was described based on material recovered fangs below a small eye; loreal absent; preocular absent; from the crop of a Dark Chanting Goshawk (Melierax one postocular; seven supralabials, with the 3rd and metabates), and the species has not been collected since 4th entering the orbit; seven infralabials, with the first (Marx, 1956; van den Berg, 2018). Specifically, such four in contact with a single pair of genials; temporal an approach is known to provide extremely valuable formula 0+1 on both sides; 19-19-17 midbody scale insights into highly cryptic and rarely sighted fossorial rows; 227 ventrals; 16+ paired subcaudals (truncated). species, such as amphisbaenids (Broadley, 1971; Shine The specimen was re-examined in mid-2019 and it was et al., 2006). These tend to be generally underrepresented discovered that the stomach was full. Upon dissection, a in museum collections and, therefore, make a case for fully intact amphisbaenian was removed (Fig. -
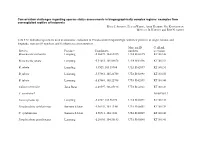
Conservation Challenges Regarding Species Status Assessments in Biogeographically Complex Regions: Examples from Overexploited Reptiles of Indonesia KYLE J
Conservation challenges regarding species status assessments in biogeographically complex regions: examples from overexploited reptiles of Indonesia KYLE J. SHANEY, ELIJAH WOSTL, AMIR HAMIDY, NIA KURNIAWAN MICHAEL B. HARVEY and ERIC N. SMITH TABLE S1 Individual specimens used in taxonomic evaluation of Pseudocalotes tympanistriga, with their province of origin, latitude and longitude, museum ID numbers, and GenBank accession numbers. Museum ID GenBank Species Province Coordinates numbers accession Bronchocela cristatella Lampung -5.36079, 104.63215 UTA R 62895 KT180148 Bronchocela jubata Lampung -5.54653, 105.04678 UTA R 62896 KT180152 B. jubata Lampung -5.5525, 105.18384 UTA R 62897 KT180151 B. jubata Lampung -5.57861, 105.22708 UTA R 62898 KT180150 B. jubata Lampung -5.57861, 105.22708 UTA R 62899 KT180146 Calotes versicolor Jawa Barat -6.49597, 106.85198 UTA R 62861 KT180149 C. versicolor* NC009683.1 Gonocephalus sp. Lampung -5.2787, 104.56198 UTA R 60571 KT180144 Pseudocalotes cybelidermus Sumatra Selatan -4.90149, 104.13401 UTA R 60551 KT180139 P. cybelidermus Sumatra Selatan -4.90711, 104.1348 UTA R 60549 KT180140 Pseudocalotes guttalineatus Lampung -5.28105, 104.56183 UTA R 60540 KT180141 P. guttalineatus Sumatra Selatan -4.90681, 104.13457 UTA R 60501 KT180142 Pseudocalotes rhammanotus Lampung -4.9394, 103.85292 MZB 10804 KT180147 Pseudocalotes species 4 Sumatra Barat -2.04294, 101.31129 MZB 13295 KT211019 Pseudocalotes tympanistriga Jawa Barat -6.74181, 107.0061 UTA R 60544 KT180143 P. tympanistriga Jawa Barat -6.74181, 107.0061 UTA R 60547 KT180145 Pogona vitticeps* AB166795.1 *Entry to GenBank by previous authors TABLE S2 Reptile species currently believed to occur Java and Sumatra, Indonesia, with IUCN Red List status, and certainty of occurrence.