DNA Transposon Expansion Is Associated with Genome Size
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Ecological Distribution of Spawning Sockeye Salmon in Three Lateral Streams, Brooks Lake, Alaska David Townsend Hoopes Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1962 Ecological distribution of spawning sockeye salmon in three lateral streams, Brooks Lake, Alaska David Townsend Hoopes Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Zoology Commons Recommended Citation Hoopes, David Townsend, "Ecological distribution of spawning sockeye salmon in three lateral streams, Brooks Lake, Alaska " (1962). Retrospective Theses and Dissertations. 2052. https://lib.dr.iastate.edu/rtd/2052 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. This dissertation has been 62-4154 microfilmed exactly as received HOOPES, David Townsend, 1932— ECO LOGICAL DISTRIBUTION OF SPAWNING SOCKEYE SALMON IN THREE LATERAL STREAMS, BROOKS LAKE, ALASKA. Iowa State University of Science and Technology Ph.D., 1962 Zoology University Microfilms, Inc., Ann Arbor, Michigan ECOLOGICAL DISTRIBUTION OF SPAWNING SOGKEYE SALMON IN THREE LATERAL STREAMS, BROOKS LAKE, ALASKA by. David Townsend Hoopes A Dissertation Submitted to the Graduate Faculty in Partial Fulfillment of The Requirements for the Degree of DOCTOR OF PHILOSOPHY c , Major Subject: Fishery Biology Approved -

Corrective Notice to the European Mudminnow (Umbra Krameri
1 Corrective notice to the European mudminnow (Umbra krameri, Walbaum 1792) 2 record from the Black Sea 3 4 5 Juraj Hajdú 1, Levente Várkonyi 2, Ján Ševc1, Tamás Müller 2*, 6 7 1 Faculty of Humanities and Natural Sciences, University of Prešov, Ul. 17. Novembra 1, 8 Prešov, Slovakia, [email protected] 9 2 Department of Aquaculture, Institute of Environmental and Landscape Management 10 Faculty of Agriculture and Environmental Science, Szent István University, 11 Páter K. u. 1, 2100 Gödöllő, Hungary, [email protected] 12 13 Abstract 14 15 Raykov et al. (2012) recorded the European mudminnow (Umbra krameri) from the Black 16 Sea, at a depth of 36.3–41 m. Morphometric comparison of the pictured specimen with 10 17 adult U. krameri and published data was conducted which excluded its taxonomic affiliation 18 to Umbridae family. 19 20 Keywords: morphometric parameters; endangered fish; taxonomic revision, 21 22 23 24 25 26 27 28 Introduction 29 30 European mudminnow (Umbra krameri) is an endemic stagnophil species of the Danube and 31 Dniester river drainages (Lelek 1987), inhabiting marshes and lowland waters densely 32 overgrown by aquatic vegetation (Wilhelm 2003, Pekárik et al. 2014). The species is 33 threatened by extinction in many of its original habitats (Simić et al. 2007). According to 34 IUCN Red List it is categorized as "Vulnerable" since its isolated and decrescent populations 35 are estimated to have declined by more than 30% in the past 10 years (Freyhof 2011). Raykov 36 et al. (2012) reported the first record of U. -

500 Natural Sciences and Mathematics
500 500 Natural sciences and mathematics Natural sciences: sciences that deal with matter and energy, or with objects and processes observable in nature Class here interdisciplinary works on natural and applied sciences Class natural history in 508. Class scientific principles of a subject with the subject, plus notation 01 from Table 1, e.g., scientific principles of photography 770.1 For government policy on science, see 338.9; for applied sciences, see 600 See Manual at 231.7 vs. 213, 500, 576.8; also at 338.9 vs. 352.7, 500; also at 500 vs. 001 SUMMARY 500.2–.8 [Physical sciences, space sciences, groups of people] 501–509 Standard subdivisions and natural history 510 Mathematics 520 Astronomy and allied sciences 530 Physics 540 Chemistry and allied sciences 550 Earth sciences 560 Paleontology 570 Biology 580 Plants 590 Animals .2 Physical sciences For astronomy and allied sciences, see 520; for physics, see 530; for chemistry and allied sciences, see 540; for earth sciences, see 550 .5 Space sciences For astronomy, see 520; for earth sciences in other worlds, see 550. For space sciences aspects of a specific subject, see the subject, plus notation 091 from Table 1, e.g., chemical reactions in space 541.390919 See Manual at 520 vs. 500.5, 523.1, 530.1, 919.9 .8 Groups of people Add to base number 500.8 the numbers following —08 in notation 081–089 from Table 1, e.g., women in science 500.82 501 Philosophy and theory Class scientific method as a general research technique in 001.4; class scientific method applied in the natural sciences in 507.2 502 Miscellany 577 502 Dewey Decimal Classification 502 .8 Auxiliary techniques and procedures; apparatus, equipment, materials Including microscopy; microscopes; interdisciplinary works on microscopy Class stereology with compound microscopes, stereology with electron microscopes in 502; class interdisciplinary works on photomicrography in 778.3 For manufacture of microscopes, see 681. -

The Rise and Fall of the Ancient Northern Pike Master Sex Determining Gene
bioRxiv preprint doi: https://doi.org/10.1101/2020.05.31.125336; this version posted June 1, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. The rise and fall of the ancient northern pike master sex determining gene Qiaowei Pan1,2, Romain Feron1,2,3, Elodie Jouanno1, Hugo Darras2, Amaury Herpin1, Ben Koop4, Eric Rondeau4, Frederick W. Goetz5, Wesley A. Larson6, Louis Bernatchez7, Mike Tringali8, Stephen S. Curran9, Eric Saillant10, Gael P.J. Denys11,12, Frank A. von Hippel13, Songlin Chen14, J. Andrés López15, Hugo Verreycken16, Konrad Ocalewicz17, Rene Guyomard18, Camille Eche19, Jerome Lluch19, Celine Roques19, Hongxia Hu20, Roger Tabor21, Patrick DeHaan21, Krista M. Nichols22, Laurent Journot23, Hugues Parrinello23, Christophe Klopp24, Elena A. Interesova25, Vladimir Trifonov26, Manfred Schartl27, John Postlethwait28, Yann Guiguen1&. &: Corresponding author. 1. INRAE, LPGP, 35000, Rennes, France. 2. Department of Ecology and Evolution, University of Lausanne,1015, Lausanne, Switzerland. 3. Swiss Institute of Bioinformatics, 1015 Lausanne, Switzerland. 4. Department of Biology, Centre for Biomedical Research, University of Victoria, Victoria, BC, V8W 3N5, Canada. 5. Environmental and Fisheries Sciences Division, Northwest Fisheries Science Center, National Marine Fisheries Service, NOAA, Seattle, WA, United States of America. 6. Fisheries Aquatic Science and Technology Laboratory at Alaska Pacific University. 4101 University Dr, Anchorage, AK 99508. 7. Institut de Biologie Intégrative et des Systèmes (IBIS), Université Laval, Québec, Québec, Canada, G1V 0A6. 8. Fish and Wildlife Conservation Commission, Florida Marine Research Institute, St. -
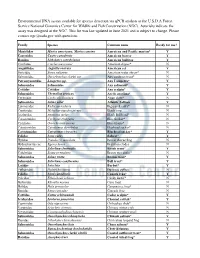
Edna Assay Development
Environmental DNA assays available for species detection via qPCR analysis at the U.S.D.A Forest Service National Genomics Center for Wildlife and Fish Conservation (NGC). Asterisks indicate the assay was designed at the NGC. This list was last updated in June 2021 and is subject to change. Please contact [email protected] with questions. Family Species Common name Ready for use? Mustelidae Martes americana, Martes caurina American and Pacific marten* Y Castoridae Castor canadensis American beaver Y Ranidae Lithobates catesbeianus American bullfrog Y Cinclidae Cinclus mexicanus American dipper* N Anguillidae Anguilla rostrata American eel Y Soricidae Sorex palustris American water shrew* N Salmonidae Oncorhynchus clarkii ssp Any cutthroat trout* N Petromyzontidae Lampetra spp. Any Lampetra* Y Salmonidae Salmonidae Any salmonid* Y Cottidae Cottidae Any sculpin* Y Salmonidae Thymallus arcticus Arctic grayling* Y Cyrenidae Corbicula fluminea Asian clam* N Salmonidae Salmo salar Atlantic Salmon Y Lymnaeidae Radix auricularia Big-eared radix* N Cyprinidae Mylopharyngodon piceus Black carp N Ictaluridae Ameiurus melas Black Bullhead* N Catostomidae Cycleptus elongatus Blue Sucker* N Cichlidae Oreochromis aureus Blue tilapia* N Catostomidae Catostomus discobolus Bluehead sucker* N Catostomidae Catostomus virescens Bluehead sucker* Y Felidae Lynx rufus Bobcat* Y Hylidae Pseudocris maculata Boreal chorus frog N Hydrocharitaceae Egeria densa Brazilian elodea N Salmonidae Salvelinus fontinalis Brook trout* Y Colubridae Boiga irregularis Brown tree snake* -

Assessment of Coastal Water Resources and Watershed Conditions at Katmai National Park and Preserve (Alaska)
National Park Service U.S. Department of the Interior Natural Resources Program Center Assessment of Coastal Water Resources and Watershed Conditions at Katmai National Park and Preserve (Alaska) Natural Resource Technical Report NPS/NRWRD/NRTR—2007/372 Cover photo: Glacier emerging from the slopes of Mt Douglas toward the Katmai coastline. August 2005. Photo: S.Nagorski 2 Assessment of Coastal Water Resources and Watershed Conditions at Katmai National Park and Preserve (Alaska) Natural Resource Technical Report NPS/NRWRD/NRTR-2007/372 Sonia Nagorski Environmental Science Program University of Alaska Southeast Juneau, AK 99801 Ginny Eckert Biology Program University of Alaska Southeast Juneau, AK 99801 Eran Hood Environmental Science Program University of Alaska Southeast Juneau, AK 99801 Sanjay Pyare Environmental Science Program University of Alaska Southeast Juneau, AK 99801 This report was prepared under Task Order J9W88050014 of the Pacific Northwest Cooperative Ecosystem Studies Unit (agreement CA90880008) Water Resources Division Natural Resource Program Center 1201 Oakridge Drive, Suite 250 Fort Collins, CO 80525 June 2007 U.S. Department of Interior Washington, D.C. 3 The Natural Resource Publication series addresses natural resource topics that are of interest and applicability to a broad readership in the National Park Service and to others in the management of natural resources, including the scientific community, the public, and the NPS conservation and environmental constituencies. Manuscripts are peer-reviewed to ensure that the information is scientifically credible, technically accurate, appropriately written for the audience, and is designed and published in a professional manner. The Natural Resource Technical Reports series is used to disseminate the peer-reviewed results of scientific studies in the physical, biological, and social sciences for both the advancement of science and the achievement of the National Park Service’s mission. -

Introduction
INTRODUCTION In nearly every body of water around the world, the most abundant vertebrate is a fi sh. From the deepest parts of the ocean to high alpine streams, fi shes live and reproduce, sometimes in places where no other vertebrates can survive. Whether peering out from a submarine while conducting deep-sea research, or stopping for a drink of water during a hike in the mountains, explorers, scientists, and naturalists fi nd fi shes. With well over 30,000 species, fi shes account for more than half of the total extant vertebrate diversity on Earth— in other words, there are more living species of fi shes than of amphibians, turtles, lizards, birds, and mammals combined. Not only are fi shes diverse in number of their species, but they are diverse in the habitats in which they live, the foods that they eat, the ways in which they reproduce, communicate, and interact with their environment, and the behaviors that they exhibit. Fishes can also be extremely abundant: the most abundant vertebrates on the planet are the small bristlemouth fi shes (Gonostomatidae) that are common throughout the vast open ocean. In some cases abundant fi shes such as cods, tunas, salmons, herrings, and anchovies support massive fi sheries that feed hundreds of millions of people. By supporting coastal communities and societies, these fi sheries (and the fi shes they target) have helped shape human history, becoming the foundation for coastal economies and an engine for global exploration and expansion. WHAT IS A FISH? Humans use the term “fi sh” to refer to several groups of vertebrates that do not have a clear set of diagnostic characteristics unique to them. -

Teleostei: Cyprinidae: Acheilognathinae) from China
Zootaxa 3790 (1): 165–176 ISSN 1175-5326 (print edition) www.mapress.com/zootaxa/ Article ZOOTAXA Copyright © 2014 Magnolia Press ISSN 1175-5334 (online edition) http://dx.doi.org/10.11646/zootaxa.3790.1.7 http://zoobank.org/urn:lsid:zoobank.org:pub:BD573A51-6656-4E86-87C2-2411443C38E5 Rhodeus albomarginatus, a new bitterling (Teleostei: Cyprinidae: Acheilognathinae) from China FAN LI1,3 & RYOICHI ARAI2 1Institute of Biodiversity Science, Ministry of Education Key Laboratory for Biodiversity Science and Ecological Engineering, Fudan University, Shanghai 200433, China. E-mail: [email protected] 2Department of Zoology, University Museum, University of Tokyo, 7–3–1 Hongo, Bunkyo-ku, Tokyo. 113-0033, Japan. E-mail: [email protected] 3Corresponding author Abstract Rhodeus albomarginatus, new species, is described from the Lvjiang River, a tributary flowing into Poyang Lake of Yang- tze River basin, in Anhui Province, China. It is distinguished from all congeneric species by unique combination of char- acters: branched dorsal-fin rays 10; branched anal-fin rays 10–11; longest simple rays of dorsal and anal fins strong and stiff, distally segmented; pelvic fin rays i 6; longitudinal scale series 34–36; transverse scale series 11; pored scales 4–7; vertebrae 33–34; colour pattern of adult males (iris black, belly reddish-orange, central part of caudal fin red, dorsal and anal fins of males edged with white margin). Key words: Cyprinidae, Rhodeus albomarginatus, new species, Yangtze River, China Introduction Bitterling belong to the subfamily Acheilognathinae in Cyprinidae and include three genera, Acheilognathus, Rhodeus and Tanakia. The genus Rhodeus can be distinguished from the other two genera by having an incomplete lateral line, no barbels, and wing-like yolk sac projections in larvae (Arai & Akai, 1988). -

Ecology and Conservation of Mudminnow Species Worldwide
FEATURE Ecology and Conservation of Mudminnow Species Worldwide Lauren M. Kuehne Ecología y conservación a nivel mundial School of Aquatic and Fishery Sciences, University of Washington, Seattle, WA 98195 de los lucios RESUMEN: en este trabajo, se revisa y resume la ecología Julian D. Olden y estado de conservación del grupo de peces comúnmente School of Aquatic and Fishery Sciences, University of Washington, Box conocido como “lucios” (anteriormente conocidos como 355020, Seattle, WA 98195, and Australian Rivers Institute, Griffith Uni- la familia Umbridae, pero recientemente reclasificados en versity, QLD, 4111, Australia. E-mail: [email protected] la Esocidae) los cuales se constituyen de sólo cinco espe- cies distribuidas en tres continentes. Estos peces de cuerpo ABSTRACT: We review and summarize the ecology and con- pequeño —que viven en hábitats de agua dulce y presentan servation status of the group of fishes commonly known as movilidad limitada— suelen presentar poblaciones aisla- “mudminnows” (formerly known as the family Umbridae but das a lo largo de distintos paisajes y son sujetos a las típi- recently reclassified as Esocidae), consisting of only five species cas amenazas que enfrentan las especies endémicas que distributed on three continents. These small-bodied fish—resid- se encuentran en contacto directo con los impactos antro- ing in freshwater habitats and exhibiting limited mobility—often pogénicos como la contaminación, alteración de hábitat occur in isolated populations across landscapes and are subject e introducción de especies no nativas. Aquí se resume el to conservation threats common to highly endemic species in conocimiento actual acerca de la distribución, relaciones close contact with anthropogenic impacts, such as pollution, filogenéticas, ecología y estado de conservación de cada habitat alteration, and nonnative species introductions. -

List of Animal Species with Ranks October 2017
Washington Natural Heritage Program List of Animal Species with Ranks October 2017 The following list of animals known from Washington is complete for resident and transient vertebrates and several groups of invertebrates, including odonates, branchipods, tiger beetles, butterflies, gastropods, freshwater bivalves and bumble bees. Some species from other groups are included, especially where there are conservation concerns. Among these are the Palouse giant earthworm, a few moths and some of our mayflies and grasshoppers. Currently 857 vertebrate and 1,100 invertebrate taxa are included. Conservation status, in the form of range-wide, national and state ranks are assigned to each taxon. Information on species range and distribution, number of individuals, population trends and threats is collected into a ranking form, analyzed, and used to assign ranks. Ranks are updated periodically, as new information is collected. We welcome new information for any species on our list. Common Name Scientific Name Class Global Rank State Rank State Status Federal Status Northwestern Salamander Ambystoma gracile Amphibia G5 S5 Long-toed Salamander Ambystoma macrodactylum Amphibia G5 S5 Tiger Salamander Ambystoma tigrinum Amphibia G5 S3 Ensatina Ensatina eschscholtzii Amphibia G5 S5 Dunn's Salamander Plethodon dunni Amphibia G4 S3 C Larch Mountain Salamander Plethodon larselli Amphibia G3 S3 S Van Dyke's Salamander Plethodon vandykei Amphibia G3 S3 C Western Red-backed Salamander Plethodon vehiculum Amphibia G5 S5 Rough-skinned Newt Taricha granulosa -

CBD Fifth National Report
Fifth National Report of Japan to the Convention on Biological Diversity Government of Japan March 2014 Contents Executive Summary 1 Chapter 1 Biodiversity: the current situation, trends and threats 7 1.1 Importance of biodiversity 7 (1) Characteristics of biodiversity in Japan from the global perspective 7 (2) Biodiversity that supports life and livelihoods 9 (3) Japan causing impacts on global biodiversity 10 (4) The economic valuation of biodiversity 11 1.2 Major changes to the biodiversity situation and trends 12 (1) The current situation of ecosystems 12 (2) The current situation of threatened wildlife 17 (3) Impacts of the Great East Japan Earthquake on biodiversity 19 1.3 The structure of the biodiversity crisis 21 (1) The four crises of biodiversity 21 (2) Japan Biodiversity Outlook (JBO) 22 1.4 The impacts of changes in biodiversity on ecosystem services, socio-economy, and culture 24 (1) Changes in the distribution of medium and large mammals and the expansion of conflicts 24 (2) Alien species 24 (3) Impacts of changes in the global environment on biodiversity 26 1.5 Future scenarios for biodiversity 28 (1) Impacts of the global warming 28 (2) The impacts of ocean acidification on coral reefs 29 (3) The forecasted expansion in the distribution of sika deer (Cervus nippon ) 30 (4) Second crisis (caused by reduced human activities) 30 Chapter 2 Implementation of the National Biodiversity Strategy and Mainstreaming Biodiversity 32 2.1 Background to the formulation of the National Biodiversity Strategy of Japan and its development -

Esox Lucius) Ecological Risk Screening Summary
Northern Pike (Esox lucius) Ecological Risk Screening Summary U.S. Fish & Wildlife Service, February 2019 Web Version, 8/26/2019 Photo: Ryan Hagerty/USFWS. Public Domain – Government Work. Available: https://digitalmedia.fws.gov/digital/collection/natdiglib/id/26990/rec/22. (February 1, 2019). 1 Native Range and Status in the United States Native Range From Froese and Pauly (2019a): “Circumpolar in fresh water. North America: Atlantic, Arctic, Pacific, Great Lakes, and Mississippi River basins from Labrador to Alaska and south to Pennsylvania and Nebraska, USA [Page and Burr 2011]. Eurasia: Caspian, Black, Baltic, White, Barents, Arctic, North and Aral Seas and Atlantic basins, southwest to Adour drainage; Mediterranean basin in Rhône drainage and northern Italy. Widely distributed in central Asia and Siberia easward [sic] to Anadyr drainage (Bering Sea basin). Historically absent from Iberian Peninsula, Mediterranean France, central Italy, southern and western Greece, eastern Adriatic basin, Iceland, western Norway and northern Scotland.” Froese and Pauly (2019a) list Esox lucius as native in Armenia, Azerbaijan, China, Georgia, Iran, Kazakhstan, Mongolia, Turkey, Turkmenistan, Uzbekistan, Albania, Austria, Belgium, Bosnia Herzegovina, Bulgaria, Croatia, Czech Republic, Denmark, Estonia, Finland, France, Germany, Greece, Hungary, Ireland, Italy, Latvia, Lithuania, Luxembourg, Macedonia, Moldova, Monaco, 1 Netherlands, Norway, Poland, Romania, Russia, Serbia, Slovakia, Slovenia, Sweden, Switzerland, United Kingdom, Ukraine, Canada, and the United States (including Alaska). From Froese and Pauly (2019a): “Occurs in Erqishi river and Ulungur lake [in China].” “Known from the Selenge drainage [in Mongolia] [Kottelat 2006].” “[In Turkey:] Known from the European Black Sea watersheds, Anatolian Black Sea watersheds, Central and Western Anatolian lake watersheds, and Gulf watersheds (Firat Nehri, Dicle Nehri).