Epigenetic Modifications in Essential Hypertension
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

The Rise and Fall of the Bovine Corpus Luteum
University of Nebraska Medical Center DigitalCommons@UNMC Theses & Dissertations Graduate Studies Spring 5-6-2017 The Rise and Fall of the Bovine Corpus Luteum Heather Talbott University of Nebraska Medical Center Follow this and additional works at: https://digitalcommons.unmc.edu/etd Part of the Biochemistry Commons, Molecular Biology Commons, and the Obstetrics and Gynecology Commons Recommended Citation Talbott, Heather, "The Rise and Fall of the Bovine Corpus Luteum" (2017). Theses & Dissertations. 207. https://digitalcommons.unmc.edu/etd/207 This Dissertation is brought to you for free and open access by the Graduate Studies at DigitalCommons@UNMC. It has been accepted for inclusion in Theses & Dissertations by an authorized administrator of DigitalCommons@UNMC. For more information, please contact [email protected]. THE RISE AND FALL OF THE BOVINE CORPUS LUTEUM by Heather Talbott A DISSERTATION Presented to the Faculty of the University of Nebraska Graduate College in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy Biochemistry and Molecular Biology Graduate Program Under the Supervision of Professor John S. Davis University of Nebraska Medical Center Omaha, Nebraska May, 2017 Supervisory Committee: Carol A. Casey, Ph.D. Andrea S. Cupp, Ph.D. Parmender P. Mehta, Ph.D. Justin L. Mott, Ph.D. i ACKNOWLEDGEMENTS This dissertation was supported by the Agriculture and Food Research Initiative from the USDA National Institute of Food and Agriculture (NIFA) Pre-doctoral award; University of Nebraska Medical Center Graduate Student Assistantship; University of Nebraska Medical Center Exceptional Incoming Graduate Student Award; the VA Nebraska-Western Iowa Health Care System Department of Veterans Affairs; and The Olson Center for Women’s Health, Department of Obstetrics and Gynecology, Nebraska Medical Center. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Differential Patterns of Allelic Loss in Estrogen Receptor-Positive Infiltrating Lobular and Ductal Breast Cancer
GENES, CHROMOSOMES & CANCER 47:1049–1066 (2008) Differential Patterns of Allelic Loss in Estrogen Receptor-Positive Infiltrating Lobular and Ductal Breast Cancer L. W. M. Loo,1 C. Ton,1,2 Y.-W. Wang,2 D. I. Grove,2 H. Bouzek,1 N. Vartanian,1 M.-G. Lin,1 X. Yuan,1 T. L. Lawton,3 J. R. Daling,2 K. E. Malone,2 C. I. Li,2 L. Hsu,2 and P.L. Porter1,2,3* 1Division of Human Biology,Fred Hutchinson Cancer Research Center,Seattle,WA 2Division of Public Health Sciences,Fred Hutchinson Cancer Research Center,Seattle,WA 3Departmentof Pathology,Universityof Washington,Seattle,WA The two main histological types of infiltrating breast cancer, lobular (ILC) and the more common ductal (IDC) carcinoma are morphologically and clinically distinct. To assess the molecular alterations associated with these breast cancer subtypes, we conducted a whole-genome study of 166 archival estrogen receptor (ER)-positive tumors (89 IDC and 77 ILC) using the Affy- metrix GeneChip® Mapping 10K Array to identify sites of loss of heterozygosity (LOH) that either distinguished, or were shared by, the two phenotypes. We found single nucleotide polymorphisms (SNPs) of high-frequency LOH (>50%) common to both ILC and IDC tumors predominately in 11q, 16q, and 17p. Overall, IDC had a slightly higher frequency of LOH events across the genome than ILC (fractional allelic loss 5 0.186 and 0.156). By comparing the average frequency of LOH by chro- mosomal arm, we found IDC tumors with significantly (P < 0.05) higher frequency of LOH on 3p, 5q, 8p, 9p, 20p, and 20q than ILC tumors. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Investigating Developmental and Functional Deficits in Neurodegenerative
UNIVERSITY OF CALIFORNIA, IRVINE Investigating developmental and functional deficits in neurodegenerative disease using transcriptomic analyses DISSERTATION submitted in partial satisfaction of the requirements for the degree of DOCTOR OF PHILOSOPHY in Biomedical Sciences by Ryan Gar-Lok Lim Dissertation Committee: Professor Leslie M. Thompson, Chair Assistant Professor Dritan Agalliu Professor Peter Donovan Professor Suzanne Sandmeyer 2016 Introduction, Figure 1.1 © 2014 Macmillan Publishers Limited. Appendix 1 © 2016 Elsevier Ltd. All other materials © 2016 Ryan Gar-Lok Lim DEDICATION This dissertation is dedicated to my parents, sister, and my wife. I love you all very much and could not have accomplished any of this without your love and support. Please take the time to reflect back on all of the moments we’ve shared, and know, that it is because of those moments I have been able to succeed. This accomplishment is as much yours as it is mine. ii TABLE OF CONTENTS Page LIST OF FIGURES vi LIST OF TABLES ix ACKNOWLEDGMENTS x CURRICULUM VITAE xiii ABSTRACT OF THE DISSERTATION xv Introduction Huntington’s disease, the neurovascular unit and the blood-brain barrier 1 1.1 Huntington’s Disease 1.2 HTT structure and function 1.2.1 Normal HTT function and possible loss-of-function contributions to HD 1.3 mHTT pathogenesis 1.3.1 The dominant pathological features of mHTT - a gain-of- toxic function? 1.3.2 Cellular pathologies and non-neuronal contributions to HD 1.4 The neurovascular unit and the blood-brain barrier 1.4.1 Structure and function -

Apoptotic Cells Inflammasome Activity During the Uptake of Macrophage
Downloaded from http://www.jimmunol.org/ by guest on September 29, 2021 is online at: average * The Journal of Immunology , 26 of which you can access for free at: 2012; 188:5682-5693; Prepublished online 20 from submission to initial decision 4 weeks from acceptance to publication April 2012; doi: 10.4049/jimmunol.1103760 http://www.jimmunol.org/content/188/11/5682 Complement Protein C1q Directs Macrophage Polarization and Limits Inflammasome Activity during the Uptake of Apoptotic Cells Marie E. Benoit, Elizabeth V. Clarke, Pedro Morgado, Deborah A. Fraser and Andrea J. Tenner J Immunol cites 56 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription http://www.jimmunol.org/content/suppl/2012/04/20/jimmunol.110376 0.DC1 This article http://www.jimmunol.org/content/188/11/5682.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2012 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 29, 2021. The Journal of Immunology Complement Protein C1q Directs Macrophage Polarization and Limits Inflammasome Activity during the Uptake of Apoptotic Cells Marie E. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Epigenetic Mechanisms Are Involved in the Oncogenic Properties of ZNF518B in Colorectal Cancer
Epigenetic mechanisms are involved in the oncogenic properties of ZNF518B in colorectal cancer Francisco Gimeno-Valiente, Ángela L. Riffo-Campos, Luis Torres, Noelia Tarazona, Valentina Gambardella, Andrés Cervantes, Gerardo López-Rodas, Luis Franco and Josefa Castillo SUPPLEMENTARY METHODS 1. Selection of genomic sequences for ChIP analysis To select the sequences for ChIP analysis in the five putative target genes, namely, PADI3, ZDHHC2, RGS4, EFNA5 and KAT2B, the genomic region corresponding to the gene was downloaded from Ensembl. Then, zoom was applied to see in detail the promoter, enhancers and regulatory sequences. The details for HCT116 cells were then recovered and the target sequences for factor binding examined. Obviously, there are not data for ZNF518B, but special attention was paid to the target sequences of other zinc-finger containing factors. Finally, the regions that may putatively bind ZNF518B were selected and primers defining amplicons spanning such sequences were searched out. Supplementary Figure S3 gives the location of the amplicons used in each gene. 2. Obtaining the raw data and generating the BAM files for in silico analysis of the effects of EHMT2 and EZH2 silencing The data of siEZH2 (SRR6384524), siG9a (SRR6384526) and siNon-target (SRR6384521) in HCT116 cell line, were downloaded from SRA (Bioproject PRJNA422822, https://www.ncbi. nlm.nih.gov/bioproject/), using SRA-tolkit (https://ncbi.github.io/sra-tools/). All data correspond to RNAseq single end. doBasics = TRUE doAll = FALSE $ fastq-dump -I --split-files SRR6384524 Data quality was checked using the software fastqc (https://www.bioinformatics.babraham. ac.uk /projects/fastqc/). The first low quality removing nucleotides were removed using FASTX- Toolkit (http://hannonlab.cshl.edu/fastxtoolkit/). -

Genetic and Genomics Laboratory Tools and Approaches
Genetic and Genomics Laboratory Tools and Approaches Meredith Yeager, PhD Cancer Genomics Research Laboratory Division of Cancer Epidemiology and Genetics [email protected] DCEG Radiation Epidemiology and Dosimetry Course 2019 www.dceg.cancer.gov/RadEpiCourse (Recent) history of genetics 2 Sequencing of the Human Genome Science 291, 1304-1351 (2001) 3 The Human Genome – 2019 • ~3.3 billion bases (A, C, G, T) • ~20,000 protein-coding genes, many non-coding RNAs (~2% of the genome) • Annotation ongoing – the initial sequencing in 2001 is still being refined, assembled and annotated, even now – hg38 • Variation (polymorphism) present within humans – Population-specific – Cosmopolitan 4 Types of polymorphisms . Single nucleotide polymorphisms (SNPs) . Common SNPs are defined as > 5% in at least one population . Abundant in genome (~50 million and counting) ATGGAACGA(G/C)AGGATA(T/A)TACGCACTATGAAG(C/A)CGGTGAGAGG . Repeats of DNA (long, short, complex, simple), insertions/deletions . A small fraction of SNPs and other types of variation are very or slightly deleterious and may contribute by themselves or with other genetic or environmental factors to a phenotype or disease 5 Different mutation rates at the nucleotide level Mutation type Mutation rate (per generation) Transition on a CpG 1.6X10-7 Transversion on a CpG 4.4X10-8 Transition: purine to purine Transition out of CpG 1.2X10-8 Transversion: purine to pyrimidine Transversion out of CpG 5.5X10-9 Substitution (average) 2.3X10-8 A and G are purines Insertion/deletion (average) 2.3X10-9 C and T are pyrimidines Mutation rate (average) 2.4X10-8 . Size of haploid genome : 3.3X109 nucleotides . -

Histone Macroh2a1.2 Promotes Metabolic Health and Leanness By
Pazienza et al. Epigenetics & Chromatin (2016) 9:45 DOI 10.1186/s13072-016-0098-9 Epigenetics & Chromatin RESEARCH Open Access Histone macroH2A1.2 promotes metabolic health and leanness by inhibiting adipogenesis Valerio Pazienza1*, Concetta Panebianco1, Francesca Rappa2,3,4, Domenico Memoli5, Michela Borghesan1,6, Sara Cannito1, Asami Oji7, Giuseppe Mazza6, Domenico Tamburrino8, Giuseppe Fusai8, Rosario Barone2,4, Giulia Bolasco9, Francesc Villarroya10,11, Joan Villarroya10,11, Kiyotaka Hatsuzawa12, Francesco Cappello2,4, Roberta Tarallo5, Tomoko Nakanishi12,13 and Manlio Vinciguerra1,4,6,14* Abstract Background: Obesity has tremendous impact on the health systems. Its epigenetic bases are unclear. MacroH2A1 is a variant of histone H2A, present in two alternatively exon-spliced isoforms macroH2A1.1 and macroH2A1.2, regulat- ing cell plasticity and proliferation, during pluripotency and tumorigenesis. Their role in adipose tissue plasticity is unknown. Results: Here, we show evidence that macroH2A1.1 protein levels in the visceral adipose tissue of obese humans positively correlate with BMI, while macroH2A1.2 is nearly absent. We thus introduced a constitutive GFP-tagged transgene for macroH2A1.2 in mice, and we characterized their metabolic health upon being fed a standard chow diet or a high fat diet. Despite unchanged food intake, these mice exhibit lower adipose mass and improved glucose metabolism both under a chow and an obesogenic diet. In the latter regimen, transgenic mice display smaller pan- creatic islets and significantly less inflammation. MacroH2A1.2 overexpression in the mouse adipose tissue induced dramatic changes in the transcript levels of key adipogenic genes; genomic analyses comparing pre-adipocytes to mature adipocytes uncovered only minor changes in macroH2A1.2 genomic distribution upon adipogenic differ- entiation and suggested differential cooperation with transcription factors. -
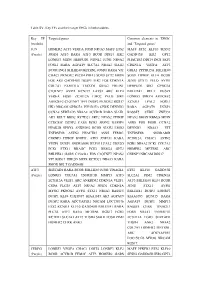
Targeted Genes Common Elements In
Table SV. Key TFs and their target DEGs in hub modules. Key TF Targeted genes Common elements in ‘DEGs’ (module) and ‘Targeted genes’ JUN HOMER2 ATF3 VEGFA FOSB NR4A3 MAFF ETS2 MAFF ETS2 KLF10 SESN2 (Purple) JOSD1 ATF3 RARA ATF3 BCOR DDIT4 IER2 GADD45B IER2 GPT2 LONRF3 MIDN HERPUD1 NDNL2 JUNB NR4A2 PHACTR3 DDIT4 ING1 SKP2 FOSL1 RARA AGPAT9 SLC7A1 NR4A2 SIAH2 CDKN1A VEGFA ATF3 BCOR ING1 BHLHE40 METRNL JOSD1 RARA VIT GRIA2 PPP1R15A BHLHE40 CHAC1 PKNOX2 PLCB4 PHF13 SOX9 SYT2 MIDN SOX9 ITPRIP KLF4 BCOR FOS AK5 GADD45B DUSP1 IER2 FOS CDKN1A JUNB STX11 PELO AVPI1 COL7A1 FAM131A TBCCD1 GRIA2 ERLIN1 HERPUD1 SIK1 GPRC5A C1QTNF7 AVPI1 KCNJ15 LATS2 ARC KLF4 BHLHE41 RELT DUSP2 VEGFA FOSB ZC3H12A LMO2 PELO SIK1 LONRF3 SPRY4 ARHGEF2 ARHGEF2 C1QTNF7 TFPI DUSP2 PKNOX2 RGS17 KCNJ15 LPAL2 FOSL1 HK1 NRCAM GPRC5A PPP1R15A CERK DENND3 RARA AGPAT9 DUSP1 CCNA2 SERTAD3 NR4A2 ACVR1B RARA SIAH1 RASSF5 CERK ZNF331 AK5 RELT BRD2 KCTD21 SKP2 NPAS2 ITPRIP NPAS2 SMOX RBM24 MIDN CCDC85C DUSP2 CARS EGR1 SESN2 RASSF5 ASNS FOS FOSB CCNA2 PIP4K2B SPRY4 ANKRD52 BCOR SIAH2 LMO2 DENND3 NR4A3 VIT TNFRSF1B ASTN2 PHACTR3 ASNS FEM1C TNFRSF1B SNORA80B CSRNP3 ITPRIP BNIP3L ATF3 ZNF331 RARA ZC3H12A CHAC1 ASTN2 VEZF1 DUSP1 SNORA80B KLF10 LPAL2 UBE2O EGR1 NR4A2 PCK1 COL7A1 PCK1 STX11 RRAGC PCK1 RBM24 GPT2 HOMER2 METRNL ARC BHLHE41 GARS C15orf41 FOS C1QTNF7 NPAS2 CSRNP3 NRCAM RGS17 VIT RGS17 UBE2O SOX9 KCTD21 NR4A3 RARA SMOX RELT GADD45B ATF3 SERTAD1 RARA BCOR BHLHE40 JUNB TINAGL1 ETS2 KLF10 GADD45B (Purple) LONRF3 COL7A1 TNFRSF1B MMP13 ATF3 SLC2A1 PIM2 CDKN1A ZC3H12A VEZF1 ARC ANKRD52 -

Genomics 93 (2009) 449–460
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Genomics 93 (2009) 449–460 Contents lists available at ScienceDirect Genomics journal homepage: www.elsevier.com/locate/ygeno Discovery of transcriptional regulators and signaling pathways in the developing pituitary gland by bioinformatic and genomic approaches Michelle L. Brinkmeier a, Shannon W. Davis a, Piero Carninci b, James W. MacDonald c, Jun Kawai b, Debashis Ghosh e,1, Yoshihide Hayashizaki b, Robert H. Lyons d, Sally A. Camper a,⁎ a Department of Human Genetics, University of Michigan Medical School, Ann Arbor, MI, 48109-5618, USA b Omics Science Center, RIKEN Yokohama Institute, Suehiro-cho, Tsurumi-ku, Yokohama, Kanagawa 230-0045, Japan c Affymetrix and cDNA Microarray Core Facility, University of Michigan Cancer Center, Ann Arbor, MI 48109-0946, USA d Department of Biological Chemistry, University of Michigan Medical School, Ann Arbor, MI, 48109-5638, USA e Department of Biostatistics, School of Public Health, University of Michigan, 48109-2029, USA article info abstract Article history: We report a catalog of the mouse embryonic pituitary gland transcriptome consisting of five cDNA libraries Received 13 December 2007 including wild type tissue from E12.5 and E14.5, Prop1df/df mutant at E14.5, and two cDNA subtractions: E14.5 Accepted 24 November 2008 WT–E14.5 Prop1df/df and E14.5 WT–E12.5 WT. DNA sequence information is assembled into a searchable Available online 11 February 2009 database with gene ontology terms representing 12,009 expressed genes. We validated coverage of the libraries by detecting most known homeobox gene transcription factor cDNAs.