Deep Ancestral Introgression Shapes Evolutionary History of Dragonflies and Damselflies
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-
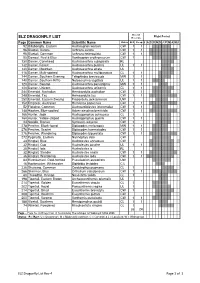
ELZ Dragonfly List Rev 4.Xlsx
Recent Flight Period ELZ DRAGONFLY LIST Records Page Common Name Scientific Name Status ELZ Co-op JASONDJFMAMJ 92 Billabongfly, Eastern Austroagrion watsoni CW 1 1 96 Bluetail, Aurora Ischnura aurora CW 1 1 96 Bluetail, Common Ischnura heterosticta CW 1 1 90 Damsel, Red & Blue Xanthagrion erythroneurum CW 1 130 Darner, Conehead Austroaeschna subapicalis RL 132 Darner, Forest Austroaeschna pulchra UL 1 1 130 Darner, Mountain Austroaeschna atrata UL 116 Darner, Multi-spotted Austroaeschna multipunctata CL 1 1 144 Darner, Southern Evening Telephlebia brevicauda MW 1 1 140 Darner, Southern Riffle Notoaeschna sagittata UL 1 1 120 Darner, Swamp Austroaeschna parvistigma MW 1 1 124 Darner, Unicorn Austroaeschna unicornis CL 1 1 244 Emerald, Australian Hemicordulia australiae CW 1 1 248 Emerald, Tau Hemicordulia tau CW 1 1 250 Emerald, Eastern Swamp Procordulia jacksoniensis UW 152 Emperor, Australian Hemianax papuensis CW 1 1 52 Flatwing, Common Austroargiolestes icteromelas CW 1 1 148 Hawker, Blue-spotted Adversaeschna brevistyla CW 1 1 166 Hunter, Jade Austrogomphus ochraceus CL 1 164 Hunter, Yellow-striped Austrogomphus guerini CW 1 1 24 Needle, Bronze Synlestes weyersii CW 1 278 Percher, Black-faced Diplacodes melanopsis MW 1 1 276 Percher, Scarlet Diplacodes haematodes CW 1 1 276 Percher, Wandering Diplacodes bipunctata CW 1 1 272 Pygmyfly, Eastern Nannophya dalei CW 34 Ringtail, Blue Austrolestes annulosus CW 32 Ringtail, Cup Austrolestes psyche UL 1 1 36 Ringtail, Iota Austrolestes io RL 32 Ringtail, Slender Austrolestes analis CW 1 1 -

The Wing Venation of Odonata
International Journal of Odonatology, 2019 Vol. 22, No. 1, 73–88, https://doi.org/10.1080/13887890.2019.1570876 The wing venation of Odonata John W. H. Trueman∗ and Richard J. Rowe Research School of Biology, Australian National University, Canberra, Australia (Received 28 July 2018; accepted 14 January 2019) Existing nomenclatures for the venation of the odonate wing are inconsistent and inaccurate. We offer a new scheme, based on the evolution and ontogeny of the insect wing and on the physical structure of wing veins, in which the veins of dragonflies and damselflies are fully reconciled with those of the other winged orders. Our starting point is the body of evidence that the insect pleuron and sternum are foreshortened leg segments and that wings evolved from leg appendages. We find that all expected longitudinal veins are present. The costa is a short vein, extending only to the nodus, and the entire costal field is sclerotised. The so-called double radial stem of Odonatoidea is a triple vein comprising the radial stem, the medial stem and the anterior cubitus, the radial and medial fields from the base of the wing to the arculus having closed when the basal sclerites fused to form a single axillary plate. In the distal part of the wing the medial and cubital fields are secondarily expanded. In Anisoptera the remnant anal field also is expanded. The dense crossvenation of Odonata, interpreted by some as an archedictyon, is secondary venation to support these expanded fields. The evolution of the odonate wing from the palaeopteran ancestor – first to the odonatoid condition, from there to the zygopteran wing in which a paddle-shaped blade is worked by two strong levers, and from there through grade Anisozygoptera to the anisopteran condition – can be simply explained. -

A Revised, ANNOTATED Checklist of Victorian Dragonflies (Insecta: Odonata)
A REVISED, ANNOTATED CHECKLIST OF Victorian DRAGONFLIES (Insecta: Odonata) I.D. ENDERS by 56 Looker Road, Montmorency, Victoria 3094, Australia E-mail: [email protected] ENDERS by , I.D. 2010. A revised, annotated checklist of the Victorian dragonflies (Insecta: Odonata). Proceedings of the Royal Society of Victoria. 122(1): 9-27. ISSN 0035-9211. Seventy-six species of Odonata are known from Victoria (26 Zygoptera; 50 Anisoptera). In the last ten years one new species Austroaeschna ingrid Theischinger, 2008 has been described from the State; Austroepigomphus praeruptus (Selys, 1857) and Pseudagrion microcephalum (Rambur, 1842) have now been recorded; and records of Rhadinosticta banksi (Tillyard, 1913) and Labidiosticta vallisi (Fraser, 1955) are judged to be erroneous. Generic names of Aeshna, and Trapezostigma have been changed. Some changes in higher level names and relationships, based on recent phylogenetic analyses, have been incorporated. Distribution maps for all species, based on museum collections, are provided. Key Words: Odonata, Zygoptera, Anisoptera, Victoria, Australia, checklist, Hemiphlebia IN the ten years since an annotated checklist of the molecular study seeks greater taxon and genome Victorian Odonata was published (Endersby 2000), sampling and, as this occurs, slow convergence be- a new species has been described from Victoria, tween the alternatives is appearing. In the meantime additional species have been recorded in the State, some framework is needed on which to list the Vic- substantial museum collection label data have be- torian fauna today. Theischinger & Endersby (2009) come available, and numerous phylogenetic stud- have tried to steer a middle course, avoiding the ex- ies have been published. Theischinger & Endersby tremes but acknowledging that change is occurring; (2009) have incorporated many of these changes into it will still annoy some but must be seen as a work in an identification guide for the adults and larvae of progress. -

Development of Encyclopedia Boyong Sleman Insekta River As Alternative Learning Resources
PROC. INTERNAT. CONF. SCI. ENGIN. ISSN 2597-5250 Volume 3, April 2020 | Pages: 629-634 E-ISSN 2598-232X Development of Encyclopedia Boyong Sleman Insekta River as Alternative Learning Resources Rini Dita Fitriani*, Sulistiyawati Biological Education Faculty of Science and Technology, UIN Sunan Kalijaga Jl. Marsda Adisucipto Yogyakarta, Indonesia Email*: [email protected] Abstract. This study aims to determine the types of insects Coleoptera, Hemiptera, Odonata, Orthoptera and Lepidoptera in the Boyong River, Sleman Regency, Yogyakarta, to develop the Encyclopedia of the Boyong River Insect and to determine the quality of the encyclopedia developed. The method used in the research inventory of the types of insects Coleoptera, Hemiptera, Odonata, Orthoptera and Lepidoptera insects in the Boyong River survey method with the results of the study found 46 species of insects consisting of 2 Coleoptera Orders, 2 Hemiptera Orders, 18 orders of Lepidoptera in Boyong River survey method with the results of the research found 46 species of insects consisting of 2 Coleoptera Orders, 2 Hemiptera Orders, 18 orders of Lepidoptera in Boyong River survey method. odonata, 4 Orthopterous Orders and 20 Lepidopterous Orders from 15 families. The encyclopedia that was developed was created using the Adobe Indesig application which was developed in printed form. Testing the quality of the encyclopedia uses a checklist questionnaire and the results of the percentage of ideals from material experts are 91.1% with very good categories, 91.7% of media experts with very good categories, peer reviewers 92.27% with very good categories, biology teachers 88, 53% with a very good category and students 89.8% with a very good category. -

Nansei Islands Biological Diversity Evaluation Project Report 1 Chapter 1
Introduction WWF Japan’s involvement with the Nansei Islands can be traced back to a request in 1982 by Prince Phillip, Duke of Edinburgh. The “World Conservation Strategy”, which was drafted at the time through a collaborative effort by the WWF’s network, the International Union for Conservation of Nature (IUCN), and the United Nations Environment Programme (UNEP), posed the notion that the problems affecting environments were problems that had global implications. Furthermore, the findings presented offered information on precious environments extant throughout the globe and where they were distributed, thereby providing an impetus for people to think about issues relevant to humankind’s harmonious existence with the rest of nature. One of the precious natural environments for Japan given in the “World Conservation Strategy” was the Nansei Islands. The Duke of Edinburgh, who was the President of the WWF at the time (now President Emeritus), naturally sought to promote acts of conservation by those who could see them through most effectively, i.e. pertinent conservation parties in the area, a mandate which naturally fell on the shoulders of WWF Japan with regard to nature conservation activities concerning the Nansei Islands. This marked the beginning of the Nansei Islands initiative of WWF Japan, and ever since, WWF Japan has not only consistently performed globally-relevant environmental studies of particular areas within the Nansei Islands during the 1980’s and 1990’s, but has put pressure on the national and local governments to use the findings of those studies in public policy. Unfortunately, like many other places throughout the world, the deterioration of the natural environments in the Nansei Islands has yet to stop. -

ANDJUS, L. & Z.ADAMOV1C, 1986. IS&Zle I Ogrozene Vrste Odonata U Siroj Okolin
OdonatologicalAbstracts 1985 NIKOLOVA & I.J. JANEVA, 1987. Tendencii v izmeneniyata na hidrobiologichnoto s’soyanie na (12331) KUGLER, J., [Ed.], 1985. Plants and animals porechieto rusenski Lom. — Tendencies in the changes Lom of the land ofIsrael: an illustrated encyclopedia, Vol. ofthe hydrobiological state of the Rusenski river 3: Insects. Ministry Defence & Soc. Prol. Nat. Israel. valley. Hidmbiologiya, Sofia 31: 65-82. (Bulg,, with 446 col. incl. ISBN 965-05-0076-6. & Russ. — Zool., Acad. Sei., pp., pis (Hebrew, Engl. s’s). (Inst. Bulg. with Engl, title & taxonomic nomenclature). Blvd Tzar Osvoboditel 1, BG-1000 Sofia). The with 48-56. Some Lists 7 odon. — Lorn R. Bul- Odon. are dealt on pp. repre- spp.; Rusenski valley, sentative described, but checklist is spp. are no pro- garia. vided. 1988 1986 (12335) KOGNITZKI, S„ 1988, Die Libellenfauna des (12332) ANDJUS, L. & Z.ADAMOV1C, 1986. IS&zle Landeskreises Erlangen-Höchstadt: Biotope, i okolini — SchrReihe ogrozene vrste Odonata u Siroj Beograda. Gefährdung, Förderungsmassnahmen. [Extinct and vulnerable Odonata species in the broader bayer. Landesaml Umweltschutz 79: 75-82. - vicinity ofBelgrade]. Sadr. Ref. 16 Skup. Ent. Jugosl, (Betzensteiner Str. 8, D-90411 Nürnberg). 16 — Hist. 41 recorded 53 localities in the VriSac, p. [abstract only]. (Serb.). (Nat. spp. were (1986) at Mus., Njegoseva 51, YU-11000 Beograd, Serbia). district, Bavaria, Germany. The fauna and the status of 27 recorded in the discussed, and During 1949-1950, spp. were area. single spp. are management measures 3 decades later, 12 spp. were not any more sighted; are suggested. they became either locally extinct or extremely rare. A list is not provided. -

The Superfamily Calopterygoidea in South China: Taxonomy and Distribution. Progress Report for 2009 Surveys Zhang Haomiao* *PH D
International Dragonfly Fund - Report 26 (2010): 1-36 1 The Superfamily Calopterygoidea in South China: taxonomy and distribution. Progress Report for 2009 surveys Zhang Haomiao* *PH D student at the Department of Entomology, College of Natural Resources and Environment, South China Agricultural University, Guangzhou 510642, China. Email: [email protected] Introduction Three families in the superfamily Calopterygoidea occur in China, viz. the Calo- pterygidae, Chlorocyphidae and Euphaeidae. They include numerous species that are distributed widely across South China, mainly in streams and upland running waters at moderate altitudes. To date, our knowledge of Chinese spe- cies has remained inadequate: the taxonomy of some genera is unresolved and no attempt has been made to map the distribution of the various species and genera. This project is therefore aimed at providing taxonomic (including on larval morphology), biological, and distributional information on the super- family in South China. In 2009, two series of surveys were conducted to Southwest China-Guizhou and Yunnan Provinces. The two provinces are characterized by karst limestone arranged in steep hills and intermontane basins. The climate is warm and the weather is frequently cloudy and rainy all year. This area is usually regarded as one of biodiversity “hotspot” in China (Xu & Wilkes, 2004). Many interesting species are recorded, the checklist and photos of these sur- veys are reported here. And the progress of the research on the superfamily Calopterygoidea is appended. Methods Odonata were recorded by the specimens collected and identified from pho- tographs. The working team includes only four people, the surveys to South- west China were completed by the author and the photographer, Mr. -

André Nel Sixtieth Anniversary Festschrift
Palaeoentomology 002 (6): 534–555 ISSN 2624-2826 (print edition) https://www.mapress.com/j/pe/ PALAEOENTOMOLOGY PE Copyright © 2019 Magnolia Press Editorial ISSN 2624-2834 (online edition) https://doi.org/10.11646/palaeoentomology.2.6.1 http://zoobank.org/urn:lsid:zoobank.org:pub:25D35BD3-0C86-4BD6-B350-C98CA499A9B4 André Nel sixtieth anniversary Festschrift DANY AZAR1, 2, ROMAIN GARROUSTE3 & ANTONIO ARILLO4 1Lebanese University, Faculty of Sciences II, Department of Natural Sciences, P.O. Box: 26110217, Fanar, Matn, Lebanon. Email: [email protected] 2State Key Laboratory of Palaeobiology and Stratigraphy, Center for Excellence in Life and Paleoenvironment, Nanjing Institute of Geology and Palaeontology, Chinese Academy of Sciences, Nanjing 210008, China. 3Institut de Systématique, Évolution, Biodiversité, ISYEB-UMR 7205-CNRS, MNHN, UPMC, EPHE, Muséum national d’Histoire naturelle, Sorbonne Universités, 57 rue Cuvier, CP 50, Entomologie, F-75005, Paris, France. 4Departamento de Biodiversidad, Ecología y Evolución, Facultad de Biología, Universidad Complutense, Madrid, Spain. FIGURE 1. Portrait of André Nel. During the last “International Congress on Fossil Insects, mainly by our esteemed Russian colleagues, and where Arthropods and Amber” held this year in the Dominican several of our members in the IPS contributed in edited volumes honoring some of our great scientists. Republic, we unanimously agreed—in the International This issue is a Festschrift to celebrate the 60th Palaeoentomological Society (IPS)—to honor our great birthday of Professor André Nel (from the ‘Muséum colleagues who have given us and the science (and still) national d’Histoire naturelle’, Paris) and constitutes significant knowledge on the evolution of fossil insects a tribute to him for his great ongoing, prolific and his and terrestrial arthropods over the years. -

The Phylogeny of the Zygopterous Dragonflies As Based on The
THE PHYLOGENY OF THE ZYGOPTEROUS DRAGON- FLIES AS BASED ON THE EVIDENCE OF THE PENES* CLARENCE HAMILTON KENNEDY, Ohio State University. This paper is merely the briefest outline of the writer's discoveries with regard to the inter-relationship of the major groups of the Zygoptera, a full account of which will appear in his thesis on the subject. Three papers1 by the writer discussing the value of this organ in classification of the Odonata have already been published. At the beginning, this study of the Zygoptera was viewed as an undertaking to define the various genera more exactly. The writer in no wise questioned the validity of the Selysian concep- tion that placed the Zygopterous subfamilies in series with the richly veined '' Calopterygines'' as primitive and the Pro- toneurinae as the latest and final reduction of venation. However, following Munz2 for the Agrioninae the writer was able to pick out here and there series of genera where the devel- opment was undoubtedly from a thinly veined wing to one richly veined, i. e., Megalagrion of Hawaii, the Argia series, Leptagrion, etc. These discoveries broke down the prejudice in the writer's mind for the irreversibility of evolution in the reduction of venation in the Odonata orders as a whole. Undoubt- ably in the Zygoptera many instances occur where a richly veined wing is merely the response to the necessity of greater wing area to support a larger body. As the study progressed the writer found almost invariably that generalized or connecting forms were usually sparsely veined as compared to their relatives. -

Dragonf Lies and Damself Lies of Europe
Dragonf lies and Damself lies of Europe A scientific approach to the identification of European Odonata without capture A simple yet detailed guide suitable both for beginners and more expert readers who wish to improve their knowledge of the order Odonata. This book contains images and photographs of all the European species having a stable population, with chapters about their anatomy, biology, behaviour, distribution range and period of flight, plus basic information about the vagrants with only a few sightings reported. On the whole, 143 reported species and over lies of Europe lies and Damself Dragonf 600 photographs are included. Published by WBA Project Srl CARLO GALLIANI, ROBERTO SCHERINI, ALIDA PIGLIA © 2017 Verona - Italy WBA Books ISSN 1973-7815 ISBN 97888903323-6-4 Supporting Institutions CONTENTS Preface 5 © WBA Project - Verona (Italy) Odonates: an introduction to the order 6 WBA HANDBOOKS 7 Dragonflies and Damselflies of Europe Systematics 7 ISSN 1973-7815 Anatomy of Odonates 9 ISBN 97888903323-6-4 Biology 14 Editorial Board: Ludivina Barrientos-Lozano, Ciudad Victoria (Mexico), Achille Casale, Sassari Mating and oviposition 23 (Italy), Mauro Daccordi, Verona (Italy), Pier Mauro Giachino, Torino (Italy), Laura Guidolin, Oviposition 34 Padova (Italy), Roy Kleukers, Leiden (Holland), Bruno Massa, Palermo (Italy), Giovanni Onore, Quito (Ecuador), Giuseppe Bartolomeo Osella, l’Aquila (Italy), Stewart B. Peck, Ottawa (Cana- Predators and preys 41 da), Fidel Alejandro Roig, Mendoza (Argentina), Jose Maria Salgado Costas, Leon (Spain), Fabio Pathogens and parasites 45 Stoch, Roma (Italy), Mauro Tretiach, Trieste (Italy), Dante Vailati, Brescia (Italy). Dichromism, androchromy and secondary homochromy 47 Editor-in-chief: Pier Mauro Giachino Particular situations in the daily life of a dragonfly 48 Managing Editor: Gianfranco Caoduro Warming up the wings 50 Translation: Alida Piglia Text revision: Michael L. -

Are Represented by 47 Spp. India’S Independence,Pp
Odonatological Abstracts 1997 mation Lanthus and abundance on sp. Cordulegastersp. and biomass is included. (14416) ALFRED, J.R.B. & A. KUMAR, 1997. Fauna 1998 ofDelhi: faunal analysis (basedon available data). Slate Fauna Ser. zool. Surv. India 6: 891-903. — (Second Author: Northern Regional Stn, Zool. Surv. India, (14420) ALFRED, A.K. DAS & A.K. Dehra Dun-248195,India). SANYAL, [Eds], 1998. [Faunal diversity in India:] Odonata. In\ J.R.B. Alfred Faunal A tabelar review of animal spp. recorded from Delhi, et al., [Eds], diversity fam. The odon. in India: commemorative volume in the 50th India;no species lists, numbers per only. a year of 172-178, ENVIS Centre, are represented by 47 spp. India’s independence,pp. Zool. Surv. India, Calcutta. — (First Author: Director, (14417) KUMAR, A., 1997. Fauna of Delhi: Odonata, Zool. Surv. India, 234/4, A.J.C. Bose Rd, Calcutta- imagos. State Fauna Ser. zool. Surv. India 6: 147-159. -700020, India). in — (Northern Regional Stn, Zool. Surv. India, Dehra The earliest reference to Indian dragonflies appears Dun-248195,India). the Sangam literature, dated prior to the 8th century and known from the A revised and updatedchecklist (47 spp.) ofthe odon. AD. At present, 449 spp. sspp. are inch 4 for the first Indian 23% of which endemic. A review fauna ofDelhi, India, spp. published territory, are and is ofthe numbers of known from various time. Precise locality data, descriptive notes presented spp. for 21 remarks onbionomy are presented spp. regions, and some considerations on conservation future studies strategies and are provided. (14418) KUMAR, A., 1997. -

Odonata) from JEZS 2014; 2 (4): 51-57 © 2014 JEZS Toebirongchhu Sub-Watershed in Punakha District, Received: 23-06-2014
Journal of Entomology and Zoology Studies 2014; 2 (4): 51-57 ISSN 2320-7078 New records of dragonflies (Odonata) from JEZS 2014; 2 (4): 51-57 © 2014 JEZS Toebirongchhu sub-watershed in Punakha District, Received: 23-06-2014 Accepted: 29-06-2014 Western Bhutan Tshering Dorji Tshering Dorji Associate Lecturer, Department of Forestry, College of Natural Resources, Royal University of Bhutan, Lobesa ABSTRACT Opportunistic survey of dragonfly diversity and distribution was done in Toebirongchhu sub-watershed within Punakha Dzongkhag, Western Bhutan to give updated list of species within the study area and the Dzongkhag, and update the species list for Bhutan. Total of 24 species belonging to 19 genera and 11 families were recorded of which 22 species are new record for the study area, 20 species for the Punakha Dzongkhag and 1 for Bhutan. The updated list of species for Punakha Dzongkhag is 28 species and for Bhutan is 85 species. Important records are Anisogomphus caudalis a Data Deficient species and a new record for Bhutan, Aristocypha (Rhinocypha) cuneata, another Data Deficient species, Epiophlebia laidlawi a Near Threatened species and Anisopleura bella a recently described species currently recorded only from Bhutan. Keywords: Dragonfly, odonata, new record, Toebirongchhu sub-watershed, Punakha, Bhutan. 1. Introduction Dragonflies belong to order Odonata and are among the most ancient of winged insects [1, 2]. The extant dragonflies are divided into two suborders, the Zygoptera or damselflies and the Anisoptera or true dragonflies, and the erstwhile third suborder Anisozygoptera is either included in Anisoptera or combined with them under the new name Epiprocta in recent times [1].