Drug Interactions with Antiretroviral Medications
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
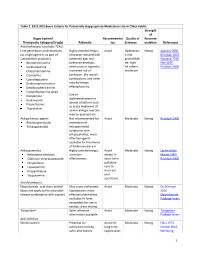
Table 2. 2012 AGS Beers Criteria for Potentially
Table 2. 2012 AGS Beers Criteria for Potentially Inappropriate Medication Use in Older Adults Strength of Organ System/ Recommendat Quality of Recomm Therapeutic Category/Drug(s) Rationale ion Evidence endation References Anticholinergics (excludes TCAs) First-generation antihistamines Highly anticholinergic; Avoid Hydroxyzin Strong Agostini 2001 (as single agent or as part of clearance reduced with e and Boustani 2007 combination products) advanced age, and promethazi Guaiana 2010 Brompheniramine tolerance develops ne: high; Han 2001 Carbinoxamine when used as hypnotic; All others: Rudolph 2008 Chlorpheniramine increased risk of moderate Clemastine confusion, dry mouth, Cyproheptadine constipation, and other Dexbrompheniramine anticholinergic Dexchlorpheniramine effects/toxicity. Diphenhydramine (oral) Doxylamine Use of diphenhydramine in Hydroxyzine special situations such Promethazine as acute treatment of Triprolidine severe allergic reaction may be appropriate. Antiparkinson agents Not recommended for Avoid Moderate Strong Rudolph 2008 Benztropine (oral) prevention of Trihexyphenidyl extrapyramidal symptoms with antipsychotics; more effective agents available for treatment of Parkinson disease. Antispasmodics Highly anticholinergic, Avoid Moderate Strong Lechevallier- Belladonna alkaloids uncertain except in Michel 2005 Clidinium-chlordiazepoxide effectiveness. short-term Rudolph 2008 Dicyclomine palliative Hyoscyamine care to Propantheline decrease Scopolamine oral secretions. Antithrombotics Dipyridamole, oral short-acting* May -

<I>Efavirenz</I>, New Therapeutic Agents for AIDS
CONFERENCE REPORTS 295 CHIMIA 1999. 53. NO.6 CONFERENCE REPORTS Chimia 53 (1999) 295-304 © Neue Schweizerische Chemische Gesellschaft ISSN 0009-4293 Second Swiss/German Meeting on Medicinal Chemistry Fruhjahrsversammlung 1999 der Neuen Schweizerischen Chemischen Gesellschaft (NSCG) 22./23. Marz 1999, Basel Vier Mini-Symposia uber Virologie, Multidrug Resistance, Immunologie und Gene Therapie Organisiert von der Sektion Medizinische Ghemie der NSGG, der Fachgruppe fUr Medizinische Chemie der GDGh und der Basler Chemischen Gesellschatt mit Unterstutzung der Pharmazeutischen Industrie. Report by the Research Team of G. Folkers' 'Correspondence: Prof. Dr. G. Folkers Department of Pharmacy Winterthurerstrasse 190 CH-8057Zi.irich E-Mail: [email protected] Discovery of Indinavir and Efavirenz, New Therapeutic Agents for AIDS Terry A. Lyle, Merck Research Laboratories, West Point, PA, USA For the treatment of HIV infection, two enzymes are of major interest: The HIV-l Protease (PR) and the Reverse- Tran- scriptase (RT). The HIV -Protease, an aspartic-acid pro- tease active as a dimer, is responsible for H 0 the cleavage of polypeptides assembled at o N~' the cell membrane. The inhibition of the Y I( ; N 1\ 0 = H protease-mediated cleavage of the viral "0 o o o precursor polyproteins results in the pro- duction of noninfectious progeny viral par- ticles. The development of lndinavir start- ed with screening a collection of renin inhibitors. A seven-amino-acid analog 1 1 which contains the hydroxyethylene tran- CONFERENCE REPORTS 296 CHIMIA 1999, 53. No.6 prevents the spread of the virus. Different nucleoside inhibitors like AZT, ddI, ddC, d4T and 3TC are already known, but new QH non-nucleoside inhibitors (NNRTI) are U'O developed to decrease the cytotoxicity and N : to improve the selectivity of the viral polymerases vs. -

Dofetilide (Tikosyn): a New Drug to Control Atrial Fibrillation
CURRENT DRUG THERAPY WALID I. SALIBA, MD Section of Cardiac Electrophysiology and Pacing, CREDIT Department of Cardiology, Cleveland Clinic Dofetilide (Tikosyn): A new drug to control atrial fibrillation ABSTRACT OFETILIDE (Tikosyn), a new antiarrhyth- mic drug, can convert atrial fibrillation Dofetilide, a new class III antiarrhythmic agent, selectively and atrial flutter to sinus rhythm in approxi- blocks a specific cardiac potassium channel, lKr, increasing mately 30% of cases and maintain sinus the effective refractory period of the myocyte and thereby rhythm after electrical or pharmacologic con- terminating reentrant arrhythmias. Given orally, it appears version for up to 1 year in 60% to 70% of to effectively convert atrial fibrillation and atrial flutter to cases, without increasing the risk of sudden sinus rhythm and maintain sinus rhythm after conversion in death in patients at high risk. appropriately selected patients. This paper reviews the Such new drugs are needed, as many of the pharmacology of dofetilide, the evidence of its antiarrhythmic drugs in use up to now have effectiveness, and the appropriate precautions in using it. actually produced higher mortality rates in clinical trials than did placebo, or cause unac- ceptable side effects. KEY POINTS This article reviews the mechanism of action, safety, effectiveness, and clinical use of Dofetilide is generally well tolerated but like other class III dofetilide. drugs can cause torsades de pointes. The risk is dose- dependent and can be minimized by adjusting the dosage • PROBLEMS WITH PREVIOUS DRUGS according to creatinine clearance and QT interval, by excluding patients with known risk factors for long QT A variety of drugs have been used to terminate syndrome and torsades de pointes, and by starting or prevent atrial and ventricular arrhythmias, treatment in an inpatient monitored setting for the first 3 but their safety, efficacy, and tolerability in days. -

Truvada (Emtricitabine / Tenofovir Disoproxil)
Pre-exposure Prophylaxis (2.3) HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use Recommended dose in HIV-1 uninfected adults: One tablet TRUVADA safely and effectively. See full prescribing information (containing 200 mg/300 mg of emtricitabine and tenofovir for TRUVADA. disoproxil fumarate) once daily taken orally with or without food. (2.3) TRUVADA® (emtricitabine/tenofovir disoproxil fumarate) tablets, for oral use Recommended dose in renally impaired HIV-uninfected Initial U.S. Approval: 2004 individuals: Do not use TRUVADA in HIV-uninfected individuals if CrCl is below 60 mL/min. If a decrease in CrCl is observed in WARNING: LACTIC ACIDOSIS/SEVERE HEPATOMEGALY WITH uninfected individuals while using TRUVADA for PrEP, evaluate STEATOSIS, POST-TREATMENT ACUTE EXACERBATION OF potential causes and re-assess potential risks and benefits of HEPATITIS B, and RISK OF DRUG RESISTANCE WITH USE OF continued use. (2.4) TRUVADA FOR PrEP IN UNDIAGNOSED HIV-1 INFECTION -----------------------DOSAGE FORMS AND STRENGTHS------------------- See full prescribing information for complete boxed warning. Tablets: 200 mg/300 mg, 167 mg/250 mg, 133 mg/200 mg, and 100 Lactic acidosis and severe hepatomegaly with steatosis, mg/150 mg of emtricitabine and tenofovir disoproxil fumarate . (3) including fatal cases, have been reported with the use of nucleoside analogs, including VIREAD, a component of TRUVADA. (5.1) --------------------------------CONTRAINDICATIONS----------------------------- TRUVADA is not approved for the treatment of chronic Do not use TRUVADA for pre-exposure prophylaxis in individuals with hepatitis B virus (HBV) infection. Severe acute unknown or positive HIV-1 status. TRUVADA should be used in exacerbations of hepatitis B have been reported in patients HIV-infected patients only in combination with other antiretroviral coinfected with HIV-1 and HBV who have discontinued agents. -

Page: Treatment-Drugs
© National HIV Curriculum PDF created September 29, 2021, 5:12 am Darunavir-Cobicistat-Tenofovir alafenamide-Emtricitabine (Symtuza) Table of Contents Darunavir-Cobicistat-Tenofovir alafenamide-Emtricitabine Symtuza Summary Drug Summary Key Clinical Trials Key Drug Interactions Drug Summary The fixed-dose combination tablet darunavir-cobicistat-tenofovir alafenamide-emtricitabine is a single-tablet regimen that can be considered for treatment-naïve or certain treatment-experienced adults living with HIV. This single-tablet regimen offers a one pill daily regimen with high barrier to resistance (due to the darunavir- cobicistat), with potentially less renal and bone toxicity as compared to regimens that include tenofovir DF; however, it has potential gastrointestinal adverse effects and drug-drug interactions, primarily due to the cobicistat component. In clinical trials, darunavir-cobicistat-tenofovir alafenamide-emtricitabine was compared to darunavir-cobicistat plus tenofovir DF-emtricitabine as initial therapy for treatment-naïve individuals and found to be equally effective in terms of viral suppression. A switch to the fixed-dose combination tablet was also compared to continuing a boosted protease inhibitor plus tenofovir DF- emtricitabine and again determined to have equivalent efficacy. The FDA has approved darunavir-cobicistat- tenofovir alafenamide-emtricitabine as a complete regimen for treatment-naïve individuals or treatment- experienced individuals who have a suppressed HIV RNA level on a stable regimen for at least 6 months and no resistance to darunavir or tenofovir. Key Clinical Trials A phase 3 trial in treatment-naïve individuals compared the fixed-dose single-tablet regimen darunavir- cobicistat-tenofovir alafenamide-emtricitabine with the regimen darunavir-cobicistat plus tenofovir DF- emtricitabine emtricitabine [AMBER]. -

Ticagrelor Antidote) by Mathematical Modeling
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Chalmers Publication Library Citation: CPT Pharmacometrics Syst. Pharmacol. (2016) 5, 313–323; doi:10.1002/psp4.12089 VC 2016 ASCPT All rights reserved ORIGINAL ARTICLE Unraveling the Pharmacokinetic Interaction of Ticagrelor and MEDI2452 (Ticagrelor Antidote) by Mathematical Modeling J Almquist1,2,3, M Penney4, S Pehrsson3, A-S Sandinge3, A Janefeldt3, S Maqbool4, S Madalli5, J Goodman4, S Nylander3 and P Gennemark3* The investigational ticagrelor-neutralizing antibody fragment, MEDI2452, is developed to rapidly and specifically reverse the antiplatelet effects of ticagrelor. However, the dynamic interaction of ticagrelor, the ticagrelor active metabolite (TAM), and MEDI2452, makes pharmacokinetic (PK) analysis nontrivial and mathematical modeling becomes essential to unravel the complex behavior of this system. We propose a mechanistic PK model, including a special observation model for post-sampling equilibration, which is validated and refined using mouse in vivo data from four studies of combined ticagrelor-MEDI2452 treatment. Model predictions of free ticagrelor and TAM plasma concentrations are subsequently used to drive a pharmacodynamic (PD) model that successfully describes platelet aggregation data. Furthermore, the model indicates that MEDI2452-bound ticagrelor is primarily eliminated together with MEDI2452 in the kidneys, and not recycled to the plasma, thereby providing a possible scenario for the extrapolation to humans. We anticipate the modeling work to improve PK and PD understanding, experimental design, and translational confidence. CPT Pharmacometrics Syst. Pharmacol. (2016) 5, 313–323; doi:10.1002/psp4.12089; published online 16 June 2016. Study Highlights WHAT IS THE CURRENT KNOWLEDGE ON THE WHAT THIS STUDY ADDS TO OUR KNOWLEDGE TOPIC? þ A mathematical model describing the simultaneous þ Antiplatelet therapy for the prevention of athero- PKs of ticagrelor and MEDI2452 in the mouse is pre- thrombotic events in patients with acute coronary syn- sented. -

Clopidogrel Compared with Other Antiplatelet Agents For
Canadian Agency for Agence canadienne Drugs and Technologies des médicaments et des in Health technologies de la santé CADTH Technology Report Issue 131 Clopidogrel Compared with Other Antiplatelet November 2010 Agents for Secondary Prevention of Vascular Events in Adults Undergoing Percutaneous Coronary Intervention: Clinical and Cost-Effectiveness Analyses Supporting Informed Decisions Until April 2006, the Canadian Agency for Drugs and Technologies in Health (CADTH) was known as the Canadian Coordinating Office for Health Technology Assessment (CCOHTA). Publications can be requested from: CADTH 600-865 Carling Avenue Ottawa ON Canada K1S 5S8 Tel: 613-226-2553 Fax: 613-226-5392 Email: [email protected] or downloaded from CADTH’s website: http://www.cadth.ca Cite as: Chen SY, Russell E, Banerjee S, Hutton B, Brown A, Asakawa K, McGahan L, Clark M, Severn M, Cox J, Sharma M. Clopidogrel Compared with Other Antiplatelet Agents for Secondary Prevention of Vascular Events in Adults Undergoing Percutaneous Coronary Intervention: Clinical and Cost-Effectiveness Analyses [Internet]. Ottawa: Canadian Agency for Drugs and Technologies in Health; 2010 (Technology report; no. 131). [cited 2010-11-01]. Available from: http://www.cadth.ca/preview.php/en/publication/2708 Production of this report is made possible by financial contributions from Health Canada and the governments of Alberta, British Columbia, Manitoba, New Brunswick, Newfoundland and Labrador, Northwest Territories, Nova Scotia, Nunavut, Prince Edward Island, Saskatchewan, and Yukon. The Canadian Agency for Drugs and Technologies in Health takes sole responsibility for the final form and content of this report. The views expressed herein do not necessarily represent the views of Health Canada or any provincial or territorial government. -

The Antimycobacterial Activity of Hypericum Perforatum Herb and the Effects of Surfactants
Utah State University DigitalCommons@USU All Graduate Theses and Dissertations Graduate Studies 8-2012 The Antimycobacterial Activity of Hypericum perforatum Herb and the Effects of Surfactants Shujie Shen Utah State University Follow this and additional works at: https://digitalcommons.usu.edu/etd Part of the Engineering Commons Recommended Citation Shen, Shujie, "The Antimycobacterial Activity of Hypericum perforatum Herb and the Effects of Surfactants" (2012). All Graduate Theses and Dissertations. 1322. https://digitalcommons.usu.edu/etd/1322 This Thesis is brought to you for free and open access by the Graduate Studies at DigitalCommons@USU. It has been accepted for inclusion in All Graduate Theses and Dissertations by an authorized administrator of DigitalCommons@USU. For more information, please contact [email protected]. i THE ANTIMYCOBACTERIAL ACTIVITY OF HYPERICUM PERFORATUM HERB AND THE EFFECTS OF SURFACTANTS by Shujie Shen A thesis submitted in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE in Biological Engineering Approved: Charles D. Miller, PhD Ronald C. Sims, PhD Major Professor Committee Member Marie K. Walsh, PhD Mark R. McLellan, PhD Committee Member Vice President for Research and Dean of the School of Graduate Studies UTAH STATE UNIVERSITY Logan, Utah 2012 ii Copyright © Shujie Shen 2012 All Rights Reserved iii ABSTRACT The Antimycobacterial Activity of Hypericum perforatum Herb and the Effects of Surfactants by Shujie Shen, Master of Science Utah State University, 2012 Major Professor: Dr. Charles D. Miller Department: Biological Engineering Due to the essential demands for novel anti-tuberculosis treatments for global tuberculosis control, this research investigated the antimycobacterial activity of Hypericum perforatum herb (commonly known as St. -

Revised Use-Function Classification (2007)
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY IPCS INTOX Data Management System (INTOX DMS) Revised Use-Function Classification (2007) The Use-Function Classification is used in two places in the INTOX Data Management System: the Communication Record and the Agent/Product Record. The two records are linked: if there is an agent record for a Centre Agent that is the subject of a call, the appropriate Intended Use-Function can be selected automatically in the Communication Record. The Use-Function Classification is used when generating reports, both standard and customized, and for searching the case and agent databases. In particular, INTOX standard reports use the top level headings of the Intended Use-Functions that were selected for Centre Agents in the Communication Record (e.g. if an agent was classified as an Analgesic for Human Use in the Communication Record, it would be logged as a Pharmaceutical for Human Use in the report). The Use-Function classification is very important for ensuring harmonized data collection. In version 4.4 of the software, 5 new additions were made to the top levels of the classification provided with the system for the classification of organisms (items XIV to XVIII). This is a 'convenience' classification to facilitate searching of the Communications database. A taxonomic classification for organisms is provided within the INTOX DMS Agent Explorer. In May/June 2006 INTOX users were surveyed to find out whether they had made any changes to the Use-Function Classification. These changes were then discussed at the 4th and 5th Meetings of INTOX Users. Version 4.5 of the INTOX DMS includes the revised pesticides classification (shown in full below). -

Download Article PDF/Slides
New Antiretrovirals in Development: Reprinted from The PRN Notebook,™ june 2002. Dr. James F. Braun, Editor-in-Chief. Tim Horn, Executive Editor. Published in New York City by the Physicians’ Research Network, Inc.,® John Graham Brown, Executive Director. For further information and other articles The View in 2002 available online, visit http://www.PRN.org All rights reserved. © june 2002. Roy “Trip” Gulick, md, mph Associate Professor of Medicine, Weill Medical College of Cornell University Director, Cornell Clinical Trials Unit, New York, New York Summary by Tim Horn Edited by Scott Hammer, md espite the fact that 16 antiretro- tiviral activity of emtricitabine was estab- Preliminary results from two random- virals are approved for use in the lished, with total daily doses of 200 mg or ized studies—FTC-302 and FTC-303—were United States, there is an indis- more producing the greatest median viral reported by Dr. Charles van der Horst and putable need for new anti-hiv com- load suppression: 1.72-1.92 log. Based on his colleagues at the 8th croi, held in Feb- pounds that have potent and these data, a once-daily dose of 200 mg ruary 2001 in Chicago (van der Horst, durable efficacy profiles, unique re- was selected for further long-term clinical 2001). FTC-302 was a blinded comparison sistance patterns, patient-friendly dosing study. “This is what we’re looking forward of emtricitabine and lamivudine, both in schedules, and minimal toxicities. To pro- to with emtricitabine,” commented Dr. combination with stavudine (Zerit) and vide prn with a glimpse of drugs current- Gulick. -

Product Monograph for CELSENTRI
PRODUCT MONOGRAPH PrCELSENTRI maraviroc Tablets 150 and 300 mg CCR5 antagonist ViiV Healthcare ULC 245, boulevard Armand-Frappier Laval, Quebec H7V 4A7 Date of Revision: July 05, 2019 Submission Control No: 226222 © 2019 ViiV Healthcare group of companies or its licensor. Trademarks are owned by or licensed to the ViiV Healthcare group of companies. Page 1 of 60 Table of Contents PART I: HEALTH PROFESSIONAL INFORMATION.........................................................3 SUMMARY PRODUCT INFORMATION ........................................................................3 INDICATIONS AND CLINICAL USE..............................................................................3 CONTRAINDICATIONS ...................................................................................................3 WARNINGS AND PRECAUTIONS..................................................................................4 ADVERSE REACTIONS....................................................................................................9 DRUG INTERACTIONS ..................................................................................................19 DOSAGE AND ADMINISTRATION..............................................................................28 OVERDOSAGE ................................................................................................................31 ACTION AND CLINICAL PHARMACOLOGY ............................................................31 STORAGE AND STABILITY..........................................................................................36 -

PATIENT INFORMATION STRIBILD® (STRY-Bild) (Elvitegravir, Cobicistat
PATIENT INFORMATION STRIBILD® (STRY-bild) (elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate) tablets Important: Ask your healthcare provider or pharmacist about medicines that should not be taken with STRIBILD. For more information, see the section “What should I tell my healthcare provider before taking STRIBILD?” What is the most important information I should know about STRIBILD? STRIBILD can cause serious side effects, including: • Worsening of Hepatitis B infection. If you have hepatitis B virus (HBV) infection and take STRIBILD, your HBV may get worse (flare-up) if you stop taking STRIBILD. A “flare-up” is when your HBV infection suddenly returns in a worse way than before. o Do not run out of STRIBILD. Refill your prescription or talk to your healthcare provider before your STRIBILD is all gone. o Do not stop taking STRIBILD without first talking to your healthcare provider. o If you stop taking STRIBILD, your healthcare provider will need to check your health often and do blood tests regularly for several months to check your HBV infection. Tell your healthcare provider about any new or unusual symptoms you may have after you stop taking STRIBILD. See “What are the possible side effects of STRIBILD?” for more information about side effects. What is STRIBILD? STRIBILD is a prescription medicine that is used without other antiretroviral medicines to treat Human Immunodeficiency Virus-1 (HIV-1) in people 12 years of age and older: • who have not received anti-HIV-1 medicines in the past, or • to replace their current anti-HIV-1 medicines: o in people who have been on the same anti-HIV-1 medicine regimen for at least 6 months, and o who have an amount of HIV-1 in their blood (this is called “viral load”) that is less than 50 copies/mL, and o have never failed past HIV-1 treatment.