A Study of the Action of Risperidone at 5-HT2A Receptors
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

TABLE 1 Studies of Antagonist Activity in Constitutively Active
TABLE 1 Studies of antagonist activity in constitutively active receptors systems shown to demonstrate inverse agonism for at least one ligand Targets are natural Gs and constitutively active mutants (CAM) of GPCRs. Of 380 antagonists, 85% of the ligands demonstrate inverse agonism. Receptor Neutral Antagonist Inverse Agonist Reference Human β2-adrenergic Dichloroisoproterenol, pindolol, labetolol, timolol, Chidiac et al., 1996; Azzi et alprenolol, propranolol, ICI 118,551, cyanopindolol al., 2001 Turkey erythrocyte β-adrenergic Propranolol, pindolol Gotze et al., 1994 Human β2-adrenergic (CAM) Propranolol Betaxolol, ICI 118,551, sotalol, timolol Samama et al., 1994; Stevens and Milligan, 1998 Human/guinea pig β1-adrenergic Atenolol, propranolol Mewes et al., 1993 Human β1-adrenergic Carvedilol CGP20712A, metoprolol, bisoprolol Engelhardt et al., 2001 Rat α2D-adrenergic Rauwolscine, yohimbine, WB 4101, idazoxan, Tian et al., 1994 phentolamine, Human α2A-adrenergic Napthazoline, Rauwolscine, idazoxan, altipamezole, levomedetomidine, Jansson et al., 1998; Pauwels MPV-2088 (–)RX811059, RX 831003 et al., 2002 Human α2C-adrenergic RX821002, yohimbine Cayla et al., 1999 Human α2D-adrenergic Prazosin McCune et al., 2000 Rat α2-adrenoceptor MK912 RX821002 Murrin et al., 2000 Porcine α2A adrenoceptor (CAM- Idazoxan Rauwolscine, yohimbine, RX821002, MK912, Wade et al., 2001 T373K) phentolamine Human α2A-adrenoceptor (CAM) Dexefaroxan, (+)RX811059, (–)RX811059, RS15385, yohimbine, Pauwels et al., 2000 atipamezole fluparoxan, WB 4101 Hamster α1B-adrenergic -

Treatment of Psychosis: 30 Years of Progress
Journal of Clinical Pharmacy and Therapeutics (2006) 31, 523–534 REVIEW ARTICLE Treatment of psychosis: 30 years of progress I. R. De Oliveira* MD PhD andM.F.Juruena MD *Department of Neuropsychiatry, School of Medicine, Federal University of Bahia, Salvador, BA, Brazil and Department of Psychological Medicine, Institute of Psychiatry, King’s College, University of London, London, UK phrenia. Thirty years ago, psychiatrists had few SUMMARY neuroleptics available to them. These were all Background: Thirty years ago, psychiatrists had compounds, today known as conventional anti- only a few choices of old neuroleptics available to psychotics, and all were liable to cause severe extra them, currently defined as conventional or typical pyramidal side-effects (EPS). Nowadays, new antipsychotics, as a result schizophrenics had to treatments are more ambitious, aiming not only to suffer the severe extra pyramidal side effects. improve psychotic symptoms, but also quality of Nowadays, new treatments are more ambitious, life and social reinsertion. We briefly but critically aiming not only to improve psychotic symptoms, outline the advances in diagnosis and treatment but also quality of life and social reinsertion. Our of schizophrenia, from the mid 1970s up to the objective is to briefly but critically review the present. advances in the treatment of schizophrenia with antipsychotics in the past 30 years. We conclude DIAGNOSIS OF SCHIZOPHRENIA that conventional antipsychotics still have a place when just the cost of treatment, a key factor in Up until the early 1970s, schizophrenia diagnoses poor regions, is considered. The atypical anti- remained debatable. The lack of uniform diagnostic psychotic drugs are a class of agents that have criteria led to relative rates of schizophrenia being become the most widely used to treat a variety of very different, for example, in New York and psychoses because of their superiority with London, as demonstrated in an important study regard to extra pyramidal symptoms. -

The In¯Uence of Medication on Erectile Function
International Journal of Impotence Research (1997) 9, 17±26 ß 1997 Stockton Press All rights reserved 0955-9930/97 $12.00 The in¯uence of medication on erectile function W Meinhardt1, RF Kropman2, P Vermeij3, AAB Lycklama aÁ Nijeholt4 and J Zwartendijk4 1Department of Urology, Netherlands Cancer Institute/Antoni van Leeuwenhoek Hospital, Plesmanlaan 121, 1066 CX Amsterdam, The Netherlands; 2Department of Urology, Leyenburg Hospital, Leyweg 275, 2545 CH The Hague, The Netherlands; 3Pharmacy; and 4Department of Urology, Leiden University Hospital, P.O. Box 9600, 2300 RC Leiden, The Netherlands Keywords: impotence; side-effect; antipsychotic; antihypertensive; physiology; erectile function Introduction stopped their antihypertensive treatment over a ®ve year period, because of side-effects on sexual function.5 In the drug registration procedures sexual Several physiological mechanisms are involved in function is not a major issue. This means that erectile function. A negative in¯uence of prescrip- knowledge of the problem is mainly dependent on tion-drugs on these mechanisms will not always case reports and the lists from side effect registries.6±8 come to the attention of the clinician, whereas a Another way of looking at the problem is drug causing priapism will rarely escape the atten- combining available data on mechanisms of action tion. of drugs with the knowledge of the physiological When erectile function is in¯uenced in a negative mechanisms involved in erectile function. The way compensation may occur. For example, age- advantage of this approach is that remedies may related penile sensory disorders may be compen- evolve from it. sated for by extra stimulation.1 Diminished in¯ux of In this paper we will discuss the subject in the blood will lead to a slower onset of the erection, but following order: may be accepted. -

Medications and Alcohol Craving
Medications and Alcohol Craving Robert M. Swift, M.D., Ph.D. The use of medications as an adjunct to alcoholism treatment is based on the premise that craving and other manifestations of alcoholism are mediated by neurobiological mechanisms. Three of the four medications approved in the United States or Europe for treating alcoholism are reported to reduce craving; these include naltrexone (ReVia™), acamprosate, and tiapride. The remaining medication, disulfiram (Antabuse®), may also possess some anticraving activity. Additional medications that have been investigated include ritanserin, which has not been shown to decrease craving or drinking levels in humans, and ondansetron, which shows promise for treating early onset alcoholics, who generally respond poorly to psychosocial treatment alone. Use of anticraving medications in combination (e.g., naltrexone plus acamprosate) may enhance their effectiveness. Future studies should address such issues as optimal dosing regimens and the development of strategies to enhance patient compliance. KEY WORDS: AOD (alcohol and other drug) craving; anti alcohol craving agents; alcohol withdrawal agents; drug therapy; neurobiological theory; alcohol cue; disulfiram; naltrexone; calcium acetylhomotaurinate; dopamine; serotonin uptake inhibitors; buspirone; treatment outcome; reinforcement; neurotransmitters; patient assessment; literature review riteria for defining alcoholism Results of craving research are often tions (i.e., pharmacotherapy) to improve vary widely. Most definitions difficult to interpret, -

Challenges and Approaches for the Development of Safer Immunomodulatory Biologics
REVIEWS Challenges and approaches for the development of safer immunomodulatory biologics Jean G. Sathish1*, Swaminathan Sethu1*, Marie-Christine Bielsky2, Lolke de Haan3, Neil S. French1, Karthik Govindappa1, James Green4, Christopher E. M. Griffiths5, Stephen Holgate6, David Jones2, Ian Kimber7, Jonathan Moggs8, Dean J. Naisbitt1, Munir Pirmohamed1, Gabriele Reichmann9, Jennifer Sims10, Meena Subramanyam11, Marque D. Todd12, Jan Willem Van Der Laan13, Richard J. Weaver14 and B. Kevin Park1 Abstract | Immunomodulatory biologics, which render their therapeutic effects by modulating or harnessing immune responses, have proven their therapeutic utility in several complex conditions including cancer and autoimmune diseases. However, unwanted adverse reactions — including serious infections, malignancy, cytokine release syndrome, anaphylaxis and hypersensitivity as well as immunogenicity — pose a challenge to the development of new (and safer) immunomodulatory biologics. In this article, we assess the safety issues associated with immunomodulatory biologics and discuss the current approaches for predicting and mitigating adverse reactions associated with their use. We also outline how these approaches can inform the development of safer immunomodulatory biologics. Immunomodulatory Biologics currently represent more than 30% of licensed The high specificity of the interactions of immu- biologics pharmaceutical products and have expanded the thera- nomodulatory biologics with their relevant immune Biotechnology-derived peutic options available -
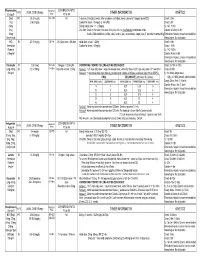
Medication Conversion Chart
Fluphenazine FREQUENCY CONVERSION RATIO ROUTE USUAL DOSE (Range) (Range) OTHER INFORMATION KINETICS Prolixin® PO to IM Oral PO 2.5-20 mg/dy QD - QID NA ↑ dose by 2.5mg/dy Q week. After symptoms controlled, slowly ↓ dose to 1-5mg/dy (dosed QD) Onset: ≤ 1hr 1mg (2-60 mg/dy) Caution for doses > 20mg/dy (↑ risk EPS) Cmax: 0.5hr 2.5mg Elderly: Initial dose = 1 - 2.5mg/dy t½: 14.7-15.3hr 5mg Oral Soln: Dilute in 2oz water, tomato or fruit juice, milk, or uncaffeinated carbonated drinks Duration of Action: 6-8hr 10mg Avoid caffeinated drinks (coffee, cola), tannics (tea), or pectinates (apple juice) 2° possible incompatibilityElimination: Hepatic to inactive metabolites 5mg/ml soln Hemodialysis: Not dialyzable HCl IM 2.5-10 mg/dy Q6-8 hr 1/3-1/2 po dose = IM dose Initial dose (usual): 1.25mg Onset: ≤ 1hr Immediate Caution for doses > 10mg/dy Cmax: 1.5-2hr Release t½: 14.7-15.3hr 2.5mg/ml Duration Action: 6-8hr Elimination: Hepatic to inactive metabolites Hemodialysis: Not dialyzable Decanoate IM 12.5-50mg Q2-3 wks 10mg po = 12.5mg IM CONVERTING FROM PO TO LONG-ACTING DECANOATE: Onset: 24-72hr (4-72hr) Long-Acting SC (12.5-100mg) (1-4 wks) Round to nearest 12.5mg Method 1: 1.25 X po daily dose = equiv decanoate dose; admin Q2-3wks. Cont ½ po daily dose X 1st few mths Cmax: 48-96hr 25mg/ml Method 2: ↑ decanoate dose over 4wks & ↓ po dose over 4-8wks as follows (accelerate taper for sx of EPS): t½: 6.8-9.6dy (single dose) ORAL DECANOATE (Administer Q 2 weeks) 15dy (14-100dy chronic administration) ORAL DOSE (mg/dy) ↓ DOSE OVER (wks) INITIAL DOSE (mg) TARGET DOSE (mg) DOSE OVER (wks) Steady State: 2mth (1.5-3mth) 5 4 6.25 6.25 0 Duration Action: 2wk (1-6wk) Elimination: Hepatic to inactive metabolites 10 4 6.25 12.5 4 Hemodialysis: Not dialyzable 20 8 6.25 12.5 4 30 8 6.25 25 4 40 8 6.25 25 4 Method 3: Admin equivalent decanoate dose Q2-3wks. -

Schizophrenia Care Guide
August 2015 CCHCS/DHCS Care Guide: Schizophrenia SUMMARY DECISION SUPPORT PATIENT EDUCATION/SELF MANAGEMENT GOALS ALERTS Minimize frequency and severity of psychotic episodes Suicidal ideation or gestures Encourage medication adherence Abnormal movements Manage medication side effects Delusions Monitor as clinically appropriate Neuroleptic Malignant Syndrome Danger to self or others DIAGNOSTIC CRITERIA/EVALUATION (PER DSM V) 1. Rule out delirium or other medical illnesses mimicking schizophrenia (see page 5), medications or drugs of abuse causing psychosis (see page 6), other mental illness causes of psychosis, e.g., Bipolar Mania or Depression, Major Depression, PTSD, borderline personality disorder (see page 4). Ideas in patients (even odd ideas) that we disagree with can be learned and are therefore not necessarily signs of schizophrenia. Schizophrenia is a world-wide phenomenon that can occur in cultures with widely differing ideas. 2. Diagnosis is made based on the following: (Criteria A and B must be met) A. Two of the following symptoms/signs must be present over much of at least one month (unless treated), with a significant impact on social or occupational functioning, over at least a 6-month period of time: Delusions, Hallucinations, Disorganized Speech, Negative symptoms (social withdrawal, poverty of thought, etc.), severely disorganized or catatonic behavior. B. At least one of the symptoms/signs should be Delusions, Hallucinations, or Disorganized Speech. TREATMENT OPTIONS MEDICATIONS Informed consent for psychotropic -

Management of Side Effects of Antipsychotics
Management of side effects of antipsychotics Oliver Freudenreich, MD, FACLP Co-Director, MGH Schizophrenia Program www.mghcme.org Disclosures I have the following relevant financial relationship with a commercial interest to disclose (recipient SELF; content SCHIZOPHRENIA): • Alkermes – Consultant honoraria (Advisory Board) • Avanir – Research grant (to institution) • Janssen – Research grant (to institution), consultant honoraria (Advisory Board) • Neurocrine – Consultant honoraria (Advisory Board) • Novartis – Consultant honoraria • Otsuka – Research grant (to institution) • Roche – Consultant honoraria • Saladax – Research grant (to institution) • Elsevier – Honoraria (medical editing) • Global Medical Education – Honoraria (CME speaker and content developer) • Medscape – Honoraria (CME speaker) • Wolters-Kluwer – Royalties (content developer) • UpToDate – Royalties, honoraria (content developer and editor) • American Psychiatric Association – Consultant honoraria (SMI Adviser) www.mghcme.org Outline • Antipsychotic side effect summary • Critical side effect management – NMS – Cardiac side effects – Gastrointestinal side effects – Clozapine black box warnings • Routine side effect management – Metabolic side effects – Motor side effects – Prolactin elevation • The man-in-the-arena algorithm www.mghcme.org Receptor profile and side effects • Alpha-1 – Hypotension: slow titration • Dopamine-2 – Dystonia: prophylactic anticholinergic – Akathisia, parkinsonism, tardive dyskinesia – Hyperprolactinemia • Histamine-1 – Sedation – Weight gain -

Appendix 13C: Clinical Evidence Study Characteristics Tables
APPENDIX 13C: CLINICAL EVIDENCE STUDY CHARACTERISTICS TABLES: PHARMACOLOGICAL INTERVENTIONS Abbreviations ............................................................................................................ 3 APPENDIX 13C (I): INCLUDED STUDIES FOR INITIAL TREATMENT WITH ANTIPSYCHOTIC MEDICATION .................................. 4 ARANGO2009 .................................................................................................................................. 4 BERGER2008 .................................................................................................................................... 6 LIEBERMAN2003 ............................................................................................................................ 8 MCEVOY2007 ................................................................................................................................ 10 ROBINSON2006 ............................................................................................................................. 12 SCHOOLER2005 ............................................................................................................................ 14 SIKICH2008 .................................................................................................................................... 16 SWADI2010..................................................................................................................................... 19 VANBRUGGEN2003 .................................................................................................................... -

(12) Patent Application Publication (10) Pub. No.: US 2011/0105536A1 Lewyn-Briscoe Et Al
US 2011 01 05536A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2011/0105536A1 Lewyn-Briscoe et al. (43) Pub. Date: May 5, 2011 (54) DOSING REGIMENASSOCATED WITH Publication Classification LONG-ACTING INUECTABLE PALIPERDONE ESTERS (51) Int. Cl. A 6LX 3/59 (2006.01) (76) Inventors: Peter H. Lewyn-Briscoe, A6IP 25/18 (2006.01) Newtown, PA (US); Cristiana Gassmann-Mayer, Pennington, NJ (US); Srihari Gopal, Belle Meade, (52) U.S. Cl. ................................................... S14/259.41 NJ (US); David W. Hough, Wallingford, PA (US); Bart M.M. Remmerie, Gent (BE); Mahesh N. (57) ABSTRACT Samtani, Flemington, NJ (US) The present application provides a method for treating (21) Appl. No.: 12/916,910 patients in need of psychiatric treatment, wherein said patient (22) Filed: Nov. 1, 2010 misses a stabilized dose of a monthly maintenance regimen of paliperidone palmitate. The present application also provides Related U.S. Application Data a method for treating psychiatric patients in need of a Switch (60) Provisional application No. 61/256,696, filed on Oct. ing treatment to paliperidone palmitate in a Sustained release 30, 2009. formulation. Patent Application Publication May 5, 2011 Sheet 1 of 6 US 2011/O105536 A1 FIG. 1 First-Order PrOCeSS Cp V CL Central (2) Zero-Order PrOCeSS Patent Application Publication May 5, 2011 Sheet 2 of 6 US 2011/O105536 A1 FIG. 2 25mgeq 50mgeq m-100mde::::: Missed doSe On WK 4. Patient returns On WK5 Missed doSe On WK 4. Patient returns On WK 6 -8-4 O 4 8 12 16 2024 -8-4 O 4 8 12 1620 24 Missed doSe On WK 4. -

Yorkshire Palliative Medicine Clinical Guidelines Group Guidelines on the Use of Antiemetics Author(S): Dr Annette Edwards (Chai
Yorkshire Palliative Medicine Clinical Guidelines Group Guidelines on the use of Antiemetics Author(s): Dr Annette Edwards (Chair) and Deborah Royle on behalf of the Yorkshire Palliative Medicine Clinical Guidelines Group Overall objective : To provide guidance on the evidence for the use of antiemetics in specialist palliative care. Search Strategy: Search strategy: Medline, Embase and Cinahl databases were searched using the words nausea, vomit$, emesis, antiemetic and drug name. Review Date: March 2008 Competing interests: None declared Disclaimer: These guidelines are the property of the Yorkshire Palliative Medicine Clinical Guidelines Group. They are intended to be used by qualified, specialist palliative care professionals as an information resource. They should be used in the clinical context of each individual patient’s needs. The clinical guidelines group takes no responsibility for any consequences of any actions taken as a result of using these guidelines. Contact Details: Dr Annette Edwards, Macmillan Consultant in Palliative Medicine, Department of Palliative Medicine, Pinderfields General Hospital, Aberford Road, Wakefield, WF1 4DG Tel: 01924 212290 E-mail: [email protected] 1 Introduction: Nausea and vomiting are common symptoms in patients with advanced cancer. A careful history, examination and appropriate investigations may help to infer the pathophysiological mechanism involved. Where possible and clinically appropriate aetiological factors should be corrected. Antiemetics are chosen based on the likely mechanism and the neurotransmitters involved in the emetic pathway. However, a recent systematic review has highlighted that evidence for the management of nausea and vomiting in advanced cancer is sparse. (Glare 2004) The following drug and non-drug treatments were reviewed to assess the strength of evidence for their use as antiemetics with particular emphasis on their use in the palliative care population. -

The Effects of Antipsychotic Treatment on Metabolic Function: a Systematic Review and Network Meta-Analysis
The effects of antipsychotic treatment on metabolic function: a systematic review and network meta-analysis Toby Pillinger, Robert McCutcheon, Luke Vano, Katherine Beck, Guy Hindley, Atheeshaan Arumuham, Yuya Mizuno, Sridhar Natesan, Orestis Efthimiou, Andrea Cipriani, Oliver Howes ****PROTOCOL**** Review questions 1. What is the magnitude of metabolic dysregulation (defined as alterations in fasting glucose, total cholesterol, low density lipoprotein (LDL) cholesterol, high density lipoprotein (HDL) cholesterol, and triglyceride levels) and alterations in body weight and body mass index associated with short-term (‘acute’) antipsychotic treatment in individuals with schizophrenia? 2. Does baseline physiology (e.g. body weight) and demographics (e.g. age) of patients predict magnitude of antipsychotic-associated metabolic dysregulation? 3. Are alterations in metabolic parameters over time associated with alterations in degree of psychopathology? 1 Searches We plan to search EMBASE, PsycINFO, and MEDLINE from inception using the following terms: 1 (Acepromazine or Acetophenazine or Amisulpride or Aripiprazole or Asenapine or Benperidol or Blonanserin or Bromperidol or Butaperazine or Carpipramine or Chlorproethazine or Chlorpromazine or Chlorprothixene or Clocapramine or Clopenthixol or Clopentixol or Clothiapine or Clotiapine or Clozapine or Cyamemazine or Cyamepromazine or Dixyrazine or Droperidol or Fluanisone or Flupehenazine or Flupenthixol or Flupentixol or Fluphenazine or Fluspirilen or Fluspirilene or Haloperidol or Iloperidone