The Heterogenization of Homogeneous Metallocene
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Opportunities for Catalysis in the 21St Century
Opportunities for Catalysis in The 21st Century A Report from the Basic Energy Sciences Advisory Committee BASIC ENERGY SCIENCES ADVISORY COMMITTEE SUBPANEL WORKSHOP REPORT Opportunities for Catalysis in the 21st Century May 14-16, 2002 Workshop Chair Professor J. M. White University of Texas Writing Group Chair Professor John Bercaw California Institute of Technology This page is intentionally left blank. Contents Executive Summary........................................................................................... v A Grand Challenge....................................................................................................... v The Present Opportunity .............................................................................................. v The Importance of Catalysis Science to DOE.............................................................. vi A Recommendation for Increased Federal Investment in Catalysis Research............. vi I. Introduction................................................................................................ 1 A. Background, Structure, and Organization of the Workshop .................................. 1 B. Recent Advances in Experimental and Theoretical Methods ................................ 1 C. The Grand Challenge ............................................................................................. 2 D. Enabling Approaches for Progress in Catalysis ..................................................... 3 E. Consensus Observations and Recommendations.................................................. -

EI-ICHI NEGISHI Herbert C
MAGICAL POWER OF TRANSITION METALS: PAST, PRESENT, AND FUTURE Nobel Lecture, December 8, 2010 by EI-ICHI NEGISHI Herbert C. Brown Laboratories of Chemistry, Purdue University, 560 Oval Drive, West Lafayette, IN 47907-2084, U.S.A. Not long ago, the primary goal of the synthesis of complex natural products and related compounds of biological and medicinal interest was to be able to synthesize them, preferably before anyone else. While this still remains a very important goal, a number of today’s top-notch synthetic chemists must feel and even think that, given ample resources and time, they are capable of synthesizing virtually all natural products and many analogues thereof. Accepting this notion, what would then be the major goals of organic synthesis in the twenty-first century? One thing appears to be unmistakably certain. Namely, we will always need, perhaps increasingly so with time, the uniquely creative field of synthetic organic and organometallic chemistry to prepare both new and existing organic compounds for the benefit and well-being of mankind. It then seems reasonably clear that, in addition to the question of what compounds to synthesize, that of how best to synthesize them will become increasingly important. As some may have said, the primary goal would then shift from aiming to be the first to synthesize a given compound to seeking its ultimately satisfactory or “last synthesis”. If one carefully goes over various aspects of organic synthetic methodology, one would soon note how primitive and limited it had been until rather recently, or perhaps even today. For the sake of argument, we may propose here that the ultimate goal of organic synthesis is “to be able to synthesize any desired and fundamentally synthesizable organic compounds (a) in high yields, (b) efficiently (in as few steps as possible, for example), (c) selectively, preferably all in t98–99% selectivity, (d) economically, and (e) safely, abbreviated hereafter as the y(es)2 manner.” with or without catalyst R1M + R2X R1R2 + MX R1, R2: carbon groups. -

Catalytic Systems Based on Cp2zrx2 (X = Cl, H), Organoaluminum
catalysts Article Catalytic Systems Based on Cp2ZrX2 (X = Cl, H), Organoaluminum Compounds and Perfluorophenylboranes: Role of Zr,Zr- and Zr,Al-Hydride Intermediates in Alkene Dimerization and Oligomerization Lyudmila V. Parfenova 1,* , Pavel V. Kovyazin 1, Almira Kh. Bikmeeva 1 and Eldar R. Palatov 2 1 Institute of Petrochemistry and Catalysis of Russian Academy of Sciences, Prospekt Oktyabrya, 141, 450075 Ufa, Russia; [email protected] (P.V.K.); [email protected] (A.K.B.) 2 Bashkir State University, st. Zaki Validi, 32, 450076 Ufa, Russia; [email protected] * Correspondence: [email protected]; Tel.: +7-347-284-3527 i i Abstract: The activity and chemoselectivity of the Cp2ZrCl2-XAlBu 2 (X = H, Bu ) and [Cp2ZrH2]2- ClAlEt2 catalytic systems activated by (Ph3C)[B(C6F5)4] or B(C6F5)3 were studied in reactions with 1-hexene. The activation of the systems by B(C6F5)3 resulted in the selective formation of head- to-tail alkene dimers in up to 93% yields. NMR studies of the reactions of Zr complexes with organoaluminum compounds (OACs) and boron activators showed the formation of Zr,Zr- and Zr,Al-hydride intermediates, for which diffusion coefficients, hydrodynamic radii, and volumes were estimated using the diffusion ordered spectroscopy DOSY. Bis-zirconium hydride clusters of type x[Cp ZrH ·Cp ZrHCl·ClAlR ]·yRnAl(C F ) − were found to be the key intermediates of alkene 2 2 2 2 6 5 3 n dimerization, whereas cationic Zr,Al-hydrides led to the formation of oligomers. Citation: Parfenova, L.V.; Kovyazin, P.V.; Bikmeeva, A.K.; Palatov, E.R. -

Organometrallic Chemistry
CHE 425: ORGANOMETALLIC CHEMISTRY SOURCE: OPEN ACCESS FROM INTERNET; Striver and Atkins Inorganic Chemistry Lecturer: Prof. O. G. Adeyemi ORGANOMETALLIC CHEMISTRY Definitions: Organometallic compounds are compounds that possess one or more metal-carbon bond. The bond must be “ionic or covalent, localized or delocalized between one or more carbon atoms of an organic group or molecule and a transition, lanthanide, actinide, or main group metal atom.” Organometallic chemistry is often described as a bridge between organic and inorganic chemistry. Organometallic compounds are very important in the chemical industry, as a number of them are used as industrial catalysts and as a route to synthesizing drugs that would not have been possible using purely organic synthetic routes. Coordinative unsaturation is a term used to describe a complex that has one or more open coordination sites where another ligand can be accommodated. Coordinative unsaturation is a very important concept in organotrasition metal chemistry. Hapticity of a ligand is the number of atoms that are directly bonded to the metal centre. Hapticity is denoted with a Greek letter η (eta) and the number of bonds a ligand has with a metal centre is indicated as a superscript, thus η1, η2, η3, ηn for hapticity 1, 2, 3, and n respectively. Bridging ligands are normally preceded by μ, with a subscript to indicate the number of metal centres it bridges, e.g. μ2–CO for a CO that bridges two metal centres. Ambidentate ligands are polydentate ligands that can coordinate to the metal centre through one or more atoms. – – – For example CN can coordinate via C or N; SCN via S or N; NO2 via N or N. -

1 the Structure and Reactivity of Single and Multiple
1 1 The Structure and Reactivity of Single and Multiple Sites on Heterogeneous and Homogeneous Catalysts: Analogies, Differences, and Challenges for Characterization Methods Adriano Zecchina , Silvia Bordiga , and Elena Groppo 1.1 Introduction The content of this book is specifi cally devoted to a description of the complexity of the catalytic centers (both homogeneous and heterogeneous) viewed as nanoma- chines for molecular assembling. Although the word “ nano ” is nowadays some- what abused, its use for catalysts science (as nanoscience) is fully justifi ed. It is a matter of fact that (i) to perform any specifi c catalytic action, the selective catalyst must necessarily possess sophisticated structure where substrates bonds are broken and formed along a specifi c path and (ii) the relevant part of this structure, usually constituted by a metal center or metal cluster surrounded by a sphere of ligands or by a solid framework or by a portion of functionalized surface, often reaches the nanometric dimension. As it will emerge from the various chapters, this vision is valid for many types of selective catalysts including catalysts for hydrogenation, polymerization, olygomerization, partial oxidation, and photocata- lytic solar energy conversion. Five chapters are devoted to the above - mentioned reactions. From the point of view of the general defi nition, homogeneous and heterogeneous selective catalysts can be treated in the same way. As homogeneous selective catalysts are concerned, the tridimensional structure surrounding the metal center can be organized with cavitand shape, while for heterogeneous cata- lysts the selectivity is the result of an accurate design and synthesis of the frame- work structures (often microporous and crystalline) where the sites are anchored. -

Cyclopentadienyl Compounds of the First Row Transition Metals And
University of Vermont ScholarWorks @ UVM Graduate College Dissertations and Theses Dissertations and Theses 2017 Cyclopentadienyl Compounds of the First Row Transition Metals and Early Actinides: Novel Main-Group Bond Forming Catalysis and New Metallacycles Justin Kane Pagano University of Vermont Follow this and additional works at: https://scholarworks.uvm.edu/graddis Part of the Chemistry Commons Recommended Citation Pagano, Justin Kane, "Cyclopentadienyl Compounds of the First Row Transition Metals and Early Actinides: Novel Main-Group Bond Forming Catalysis and New Metallacycles" (2017). Graduate College Dissertations and Theses. 700. https://scholarworks.uvm.edu/graddis/700 This Dissertation is brought to you for free and open access by the Dissertations and Theses at ScholarWorks @ UVM. It has been accepted for inclusion in Graduate College Dissertations and Theses by an authorized administrator of ScholarWorks @ UVM. For more information, please contact [email protected]. CYCLOPENTADIENYL COMPOUNDS OF THE FIRST ROW TRANSITION METALS AND EARLY ACTINIDES: NOVEL MAIN-GROUP BOND FORMING CATALYSIS AND NEW METALLACYCLES A Dissertation Presented by Justin Kane Pagano to The Faculty of the Graduate College of The University of Vermont In Partial Fulfilment of the Requirements For the Degree of Doctor of Philosophy Specializing in Chemistry May, 2017 Defense Date: November 29, 2016 Dissertation Examination Committee: Rory Waterman, Ph. D., Advisor John M. Hughes, Ph. D., Chairperson Matthias Brewer, Ph. D. Jaqueline L. Kiplinger, Ph. D. Matthew D. Liptak, Ph. D. Cynthia J. Forehand, Ph. D., Dean of the Graduate College ABSTRACT Cyclopentadienyl first row transition-metal compounds have been well studied 5 since the 1950’s, with the nearly ubiquitous CpFe(CO)2Me (FpMe) (Cp = η -C5H5) being one of the first organometallics to be fully characterized. -

Synthesis and Reactivity of Cyclopentadienyl Based Organometallic Compounds and Their Electrochemical and Biological Properties
Synthesis and reactivity of cyclopentadienyl based organometallic compounds and their electrochemical and biological properties Sasmita Mishra Department of Chemistry National Institute of Technology Rourkela Synthesis and reactivity of cyclopentadienyl based organometallic compounds and their electrochemical and biological properties Dissertation submitted to the National Institute of Technology Rourkela In partial fulfillment of the requirements of the degree of Doctor of Philosophy in Chemistry by Sasmita Mishra (Roll Number: 511CY604) Under the supervision of Prof. Saurav Chatterjee February, 2017 Department of Chemistry National Institute of Technology Rourkela Department of Chemistry National Institute of Technology Rourkela Certificate of Examination Roll Number: 511CY604 Name: Sasmita Mishra Title of Dissertation: ''Synthesis and reactivity of cyclopentadienyl based organometallic compounds and their electrochemical and biological properties We the below signed, after checking the dissertation mentioned above and the official record book(s) of the student, hereby state our approval of the dissertation submitted in partial fulfillment of the requirements of the degree of Doctor of Philosophy in Chemistry at National Institute of Technology Rourkela. We are satisfied with the volume, quality, correctness, and originality of the work. --------------------------- Prof. Saurav Chatterjee Principal Supervisor --------------------------- --------------------------- Prof. A. Sahoo. Prof. G. Hota Member (DSC) Member (DSC) --------------------------- -

Heterogeneous Catalytic Oligomerization of Ethylene
Heterogeneous Catalytic Oligomerization of Ethylene Oliver Dennis Jan A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Washington 2017 Reading Committee: Fernando Resende, Chair Rick Gustafson Anthony Dichiara Program Authorized to Offer Degree: School of Environmental and Forest Sciences © Copyright 2017 Oliver Dennis Jan ii University of Washington Abstract Heterogeneous Catalytic Oligomerization of Ethylene Oliver Dennis Jan Chair of the Supervisory Committee: Assistant Professor Fernando Resende School of Environmental and Forest Sciences Throughout this work, we report results for the oligomerization of ethylene over Ni-Hβ in a packed bed reactor. We performed a parameterized study over temperature (30ºC-190ºC), pressure (8.5-25.6 bar), and weighted hourly space velocity (2.0-5.5 hr-1). We observed that the ethylene conversion increased with reaction pressure due primarily to the slower velocities at higher pressures. Increasing the temperature of the reactor led to the formation of larger oligomers and coke, but its effect on the conversion was small. The space velocity played an important role on ethylene conversion and product selectivity, with higher conversions observed at lower space velocities and higher selectivities to butene at higher space velocities. We also conducted a long experiment to determine the activity of the Ni-Hβ catalyst over 72 hours-on-stream at 19.0 bar partial pressure of ethylene, 120ºC, and 3.1 hr-1 WHSV. We observed that catalyst deactivation occurred only during the startup period largely due to coke formation. Despite this initial iii deactivation, negligible coke formation occurred after 8 hours time-on-stream, as the conversion remained steady at 47% for the duration of the experiment. -

US20100160506A1.Pdf
US 2010.0160506A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2010/0160506 A1 Wu et al. (43) Pub. Date: Jun. 24, 2010 (54) PRODUCTION OFSYNTHETIC Publication Classification HYDROCARBON FLUIDS, PLASTICIZERS (51) Int. Cl. AND SYNTHETICLUBRICANT BASE CSK 5/55 (2006.01) STOCKS FROM RENEWABLE FEEDSTOCKS CSK 5/109 (2006.01) C07D 30/03 (2006.01) (76) Inventors: Margaret May-Som Wu, Skillman, C07D 31 7/24 (2006.01) NJ (US); Karla Schall Colle, C07C I/207 (2006.01) Houston, TX (US); Ramzi Yanni (52) U.S. Cl. ......... 524/114; 524/280; 549/523: 549/229; Saleh, Baton Rouge, LA (US); 585/327 Allen D. Godwin, Seabrook, TX (57) ABSTRACT (US); John Edmond Randolph This disclosure is directed to an integrated method for making Stanat, Houston, TX (US) synthetic hydrocarbon fluids, plasticizers and polar synthetic lubricant base stocks from a renewable feedstock. More par Correspondence Address: ticularly, the disclosure is directed to a metathesis reaction of ExxonMobil Research & Engineering Company natural oil or its derivative ester and ethylene in the presence P.O. Box 900, 1545 Route 22 East of an effective amount of a metathesis catalyst to form linear Annandale, NJ 08801-0900 (US) alpha-olefins, internal olefins and reduced chain length trig lycerides. The linear alpha-olefins and/or internal olefins are polymerized to produce synthetic hydrocarbon fluids in the (21) Appl. No.: 12/633,742 presence of a suitable catalyst. The reduced chain length triglycerides are converted into polar synthetic lubricant base (22) Filed: Dec. 17, 2009 stocks or plasticizers by hydrogenation, isomerization, fol lowed by hydrogenations, or by hydroisomerization pro cesses. -

Basic Research Needs for Catalysis Science
Basic Research Needs for Catalysis Science Report of the Basic Energy Sciences Workshop on Basic Research Needs for Catalysis Science to Transform Energy Technologies May 8–10, 2017 Image courtesy of Argonne National Laboratory. DISCLAIMER This report was prepared as an account of a workshop sponsored by the U.S. Department of Energy. Neither the United States Government nor any agency thereof, nor any of their employees or officers, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof. The views and opinions of document authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof. Copyrights to portions of this report (including graphics) are reserved by original copyright holders or their assignees, and are used by the Government’s license and by permission. Requests to use any images must be made to the provider identified in the image credits. This report is available in pdf format at https://science.energy.gov/bes/community-resources/reports/ REPORT OF THE BASIC RESEARCH NEEDS WORKSHOP FOR CATALYSIS SCIENCE Basic Research Needs for Catalysis Science TO TRANSFORM ENERGY TECHNOLOGIES Report from the U.S. Department of Energy, Office of Basic Energy Sciences Workshop May 8–10, 2017, in Gaithersburg, Maryland CHAIR: ASSOCIATE CHAIRS: Carl A. -

Low Cost/Waste Catalyst for Fatty Acid Methyl Ester Production
Article Number: 2AEBF76 A Paper presented at the 39th CSN Annual International Conference, Workshop and Exhibition, Rivers State University of Science and Technology, Port Harcourt, Nigeria. 18th – 23rd September 2016 Copyright ©2018 Author(s) retain the copyright of this article Conference Proceedings http://www.proceedings.academicjournals.org/ Full Length Research Paper Low cost/waste catalyst for fatty acid methyl ester production M. O. Ekeoma1*, P. A. C. Okoye2 and V. I. E. Ajiwe2 1Department of Chemistry, College of Physical and Applied Sciences, Michael Okpara University of Agriculture, Umudike, P. M. B. 7267, Umuahia, Abia State, Nigeria. 2Department of Pure and Industrial Chemistry, Faculty of Physical Sciences, Nnamdi Azikiwe University, Awka, Anambra State, Nigeria. Non-edible crude karanj (Pongamia pinnata) oil (CKO) with high free fatty acid (FFA) content was used as effective renewable feedstock for fatty acid methyl ester (FAME) production. Calcium feldspar clay, a rare compositional variety of plagioclase clay, a low cost, abundant earth resource, containing over 90% CaO and belonging to the class of anorthite clay was used as heterogeneous catalyst in direct conversion of high FFA crude karanj oil to fatty acid methyl esters. The efficiency of the catalyst was made possible by the structural rearrangement of the mixed metal oxides' content of the catalyst at prolonged high temperatures, a behaviour characteristic of glass transitions and properties of amorphous phases of plagioclase feldspar, and thus was transformed into solid acid particles such as acidic mesoporous aluminium silicate mixed oxides. Optimum FAME yield of 98.97% was obtained at 4 h reaction time, 6 wt% catalyst loading, 9:1 methanol to CKO molar ratio and at methanol reflux temperature. -
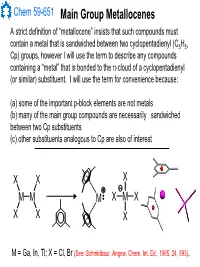
Main Group Metallocenes
Chem 59-651 Main Group Metallocenes A strict definition of “metallocene” insists that such compounds must contain a metal that is sandwiched between two cyclopentadienyl (C5H5, Cp) groups, however I will use the term to describe any compounds containing a “metal” that is bonded to the π-cloud of a cyclopentadienyl (or similar) substituent. I will use the term for convenience because: (a) some of the important p-block elements are not metals (b) many of the main group compounds are necessarily sandwiched between two Cp substituents (c) other substituents analogous to Cp are also of interest XX X MM M XMX X X X M = Ga, In, Tl; X = Cl, Br (See: Schmidbaur, Angew. Chem. Int. Ed., 1985, 24, 893). Chem 59-651 Cyclopentadienyl Ligand Basics The structural features and bonding interactions between cyclopentadienyl groups and metal atoms shows considerable variation and is described using a few different conventions (See Jutzi and Burford, Chem. Rev., 1999, 99, 969 and references therein). In most main group metallocenes, even those that are σ-bonded in the solid state, the rings rotate rapidly through a series of 1,2-shifts. A Cp group can be considered to be p-bonded to an element if the Hapticity: element sits inside the cylinder defined by the 5 carbon atoms of M MM the Cp ligand. The center of mass of the 5 C atoms is known as the centroid. η1-Cp η2-Cp η3-Cp M M M A σ-bonded Cp can be identified η4-Cp η5-Cp ≡η5-Cp by the angle at the ipso carbon M and bond lengths should indicate single bonds to the ipso carbon and a localized diene structure for σ the α and β carbon fragment.