By Makena L. Clive a Dissertation Submitted to Johns Hopkins University in Conformity with the Requirements for the Degree of Do
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Rise and Fall of the Bovine Corpus Luteum
University of Nebraska Medical Center DigitalCommons@UNMC Theses & Dissertations Graduate Studies Spring 5-6-2017 The Rise and Fall of the Bovine Corpus Luteum Heather Talbott University of Nebraska Medical Center Follow this and additional works at: https://digitalcommons.unmc.edu/etd Part of the Biochemistry Commons, Molecular Biology Commons, and the Obstetrics and Gynecology Commons Recommended Citation Talbott, Heather, "The Rise and Fall of the Bovine Corpus Luteum" (2017). Theses & Dissertations. 207. https://digitalcommons.unmc.edu/etd/207 This Dissertation is brought to you for free and open access by the Graduate Studies at DigitalCommons@UNMC. It has been accepted for inclusion in Theses & Dissertations by an authorized administrator of DigitalCommons@UNMC. For more information, please contact [email protected]. THE RISE AND FALL OF THE BOVINE CORPUS LUTEUM by Heather Talbott A DISSERTATION Presented to the Faculty of the University of Nebraska Graduate College in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy Biochemistry and Molecular Biology Graduate Program Under the Supervision of Professor John S. Davis University of Nebraska Medical Center Omaha, Nebraska May, 2017 Supervisory Committee: Carol A. Casey, Ph.D. Andrea S. Cupp, Ph.D. Parmender P. Mehta, Ph.D. Justin L. Mott, Ph.D. i ACKNOWLEDGEMENTS This dissertation was supported by the Agriculture and Food Research Initiative from the USDA National Institute of Food and Agriculture (NIFA) Pre-doctoral award; University of Nebraska Medical Center Graduate Student Assistantship; University of Nebraska Medical Center Exceptional Incoming Graduate Student Award; the VA Nebraska-Western Iowa Health Care System Department of Veterans Affairs; and The Olson Center for Women’s Health, Department of Obstetrics and Gynecology, Nebraska Medical Center. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Differential Patterns of Allelic Loss in Estrogen Receptor-Positive Infiltrating Lobular and Ductal Breast Cancer
GENES, CHROMOSOMES & CANCER 47:1049–1066 (2008) Differential Patterns of Allelic Loss in Estrogen Receptor-Positive Infiltrating Lobular and Ductal Breast Cancer L. W. M. Loo,1 C. Ton,1,2 Y.-W. Wang,2 D. I. Grove,2 H. Bouzek,1 N. Vartanian,1 M.-G. Lin,1 X. Yuan,1 T. L. Lawton,3 J. R. Daling,2 K. E. Malone,2 C. I. Li,2 L. Hsu,2 and P.L. Porter1,2,3* 1Division of Human Biology,Fred Hutchinson Cancer Research Center,Seattle,WA 2Division of Public Health Sciences,Fred Hutchinson Cancer Research Center,Seattle,WA 3Departmentof Pathology,Universityof Washington,Seattle,WA The two main histological types of infiltrating breast cancer, lobular (ILC) and the more common ductal (IDC) carcinoma are morphologically and clinically distinct. To assess the molecular alterations associated with these breast cancer subtypes, we conducted a whole-genome study of 166 archival estrogen receptor (ER)-positive tumors (89 IDC and 77 ILC) using the Affy- metrix GeneChip® Mapping 10K Array to identify sites of loss of heterozygosity (LOH) that either distinguished, or were shared by, the two phenotypes. We found single nucleotide polymorphisms (SNPs) of high-frequency LOH (>50%) common to both ILC and IDC tumors predominately in 11q, 16q, and 17p. Overall, IDC had a slightly higher frequency of LOH events across the genome than ILC (fractional allelic loss 5 0.186 and 0.156). By comparing the average frequency of LOH by chro- mosomal arm, we found IDC tumors with significantly (P < 0.05) higher frequency of LOH on 3p, 5q, 8p, 9p, 20p, and 20q than ILC tumors. -

Apoptotic Cells Inflammasome Activity During the Uptake of Macrophage
Downloaded from http://www.jimmunol.org/ by guest on September 29, 2021 is online at: average * The Journal of Immunology , 26 of which you can access for free at: 2012; 188:5682-5693; Prepublished online 20 from submission to initial decision 4 weeks from acceptance to publication April 2012; doi: 10.4049/jimmunol.1103760 http://www.jimmunol.org/content/188/11/5682 Complement Protein C1q Directs Macrophage Polarization and Limits Inflammasome Activity during the Uptake of Apoptotic Cells Marie E. Benoit, Elizabeth V. Clarke, Pedro Morgado, Deborah A. Fraser and Andrea J. Tenner J Immunol cites 56 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription http://www.jimmunol.org/content/suppl/2012/04/20/jimmunol.110376 0.DC1 This article http://www.jimmunol.org/content/188/11/5682.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2012 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 29, 2021. The Journal of Immunology Complement Protein C1q Directs Macrophage Polarization and Limits Inflammasome Activity during the Uptake of Apoptotic Cells Marie E. -

Genetic and Genomics Laboratory Tools and Approaches
Genetic and Genomics Laboratory Tools and Approaches Meredith Yeager, PhD Cancer Genomics Research Laboratory Division of Cancer Epidemiology and Genetics [email protected] DCEG Radiation Epidemiology and Dosimetry Course 2019 www.dceg.cancer.gov/RadEpiCourse (Recent) history of genetics 2 Sequencing of the Human Genome Science 291, 1304-1351 (2001) 3 The Human Genome – 2019 • ~3.3 billion bases (A, C, G, T) • ~20,000 protein-coding genes, many non-coding RNAs (~2% of the genome) • Annotation ongoing – the initial sequencing in 2001 is still being refined, assembled and annotated, even now – hg38 • Variation (polymorphism) present within humans – Population-specific – Cosmopolitan 4 Types of polymorphisms . Single nucleotide polymorphisms (SNPs) . Common SNPs are defined as > 5% in at least one population . Abundant in genome (~50 million and counting) ATGGAACGA(G/C)AGGATA(T/A)TACGCACTATGAAG(C/A)CGGTGAGAGG . Repeats of DNA (long, short, complex, simple), insertions/deletions . A small fraction of SNPs and other types of variation are very or slightly deleterious and may contribute by themselves or with other genetic or environmental factors to a phenotype or disease 5 Different mutation rates at the nucleotide level Mutation type Mutation rate (per generation) Transition on a CpG 1.6X10-7 Transversion on a CpG 4.4X10-8 Transition: purine to purine Transition out of CpG 1.2X10-8 Transversion: purine to pyrimidine Transversion out of CpG 5.5X10-9 Substitution (average) 2.3X10-8 A and G are purines Insertion/deletion (average) 2.3X10-9 C and T are pyrimidines Mutation rate (average) 2.4X10-8 . Size of haploid genome : 3.3X109 nucleotides . -

Histone Macroh2a1.2 Promotes Metabolic Health and Leanness By
Pazienza et al. Epigenetics & Chromatin (2016) 9:45 DOI 10.1186/s13072-016-0098-9 Epigenetics & Chromatin RESEARCH Open Access Histone macroH2A1.2 promotes metabolic health and leanness by inhibiting adipogenesis Valerio Pazienza1*, Concetta Panebianco1, Francesca Rappa2,3,4, Domenico Memoli5, Michela Borghesan1,6, Sara Cannito1, Asami Oji7, Giuseppe Mazza6, Domenico Tamburrino8, Giuseppe Fusai8, Rosario Barone2,4, Giulia Bolasco9, Francesc Villarroya10,11, Joan Villarroya10,11, Kiyotaka Hatsuzawa12, Francesco Cappello2,4, Roberta Tarallo5, Tomoko Nakanishi12,13 and Manlio Vinciguerra1,4,6,14* Abstract Background: Obesity has tremendous impact on the health systems. Its epigenetic bases are unclear. MacroH2A1 is a variant of histone H2A, present in two alternatively exon-spliced isoforms macroH2A1.1 and macroH2A1.2, regulat- ing cell plasticity and proliferation, during pluripotency and tumorigenesis. Their role in adipose tissue plasticity is unknown. Results: Here, we show evidence that macroH2A1.1 protein levels in the visceral adipose tissue of obese humans positively correlate with BMI, while macroH2A1.2 is nearly absent. We thus introduced a constitutive GFP-tagged transgene for macroH2A1.2 in mice, and we characterized their metabolic health upon being fed a standard chow diet or a high fat diet. Despite unchanged food intake, these mice exhibit lower adipose mass and improved glucose metabolism both under a chow and an obesogenic diet. In the latter regimen, transgenic mice display smaller pan- creatic islets and significantly less inflammation. MacroH2A1.2 overexpression in the mouse adipose tissue induced dramatic changes in the transcript levels of key adipogenic genes; genomic analyses comparing pre-adipocytes to mature adipocytes uncovered only minor changes in macroH2A1.2 genomic distribution upon adipogenic differ- entiation and suggested differential cooperation with transcription factors. -
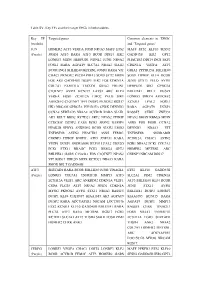
Targeted Genes Common Elements In
Table SV. Key TFs and their target DEGs in hub modules. Key TF Targeted genes Common elements in ‘DEGs’ (module) and ‘Targeted genes’ JUN HOMER2 ATF3 VEGFA FOSB NR4A3 MAFF ETS2 MAFF ETS2 KLF10 SESN2 (Purple) JOSD1 ATF3 RARA ATF3 BCOR DDIT4 IER2 GADD45B IER2 GPT2 LONRF3 MIDN HERPUD1 NDNL2 JUNB NR4A2 PHACTR3 DDIT4 ING1 SKP2 FOSL1 RARA AGPAT9 SLC7A1 NR4A2 SIAH2 CDKN1A VEGFA ATF3 BCOR ING1 BHLHE40 METRNL JOSD1 RARA VIT GRIA2 PPP1R15A BHLHE40 CHAC1 PKNOX2 PLCB4 PHF13 SOX9 SYT2 MIDN SOX9 ITPRIP KLF4 BCOR FOS AK5 GADD45B DUSP1 IER2 FOS CDKN1A JUNB STX11 PELO AVPI1 COL7A1 FAM131A TBCCD1 GRIA2 ERLIN1 HERPUD1 SIK1 GPRC5A C1QTNF7 AVPI1 KCNJ15 LATS2 ARC KLF4 BHLHE41 RELT DUSP2 VEGFA FOSB ZC3H12A LMO2 PELO SIK1 LONRF3 SPRY4 ARHGEF2 ARHGEF2 C1QTNF7 TFPI DUSP2 PKNOX2 RGS17 KCNJ15 LPAL2 FOSL1 HK1 NRCAM GPRC5A PPP1R15A CERK DENND3 RARA AGPAT9 DUSP1 CCNA2 SERTAD3 NR4A2 ACVR1B RARA SIAH1 RASSF5 CERK ZNF331 AK5 RELT BRD2 KCTD21 SKP2 NPAS2 ITPRIP NPAS2 SMOX RBM24 MIDN CCDC85C DUSP2 CARS EGR1 SESN2 RASSF5 ASNS FOS FOSB CCNA2 PIP4K2B SPRY4 ANKRD52 BCOR SIAH2 LMO2 DENND3 NR4A3 VIT TNFRSF1B ASTN2 PHACTR3 ASNS FEM1C TNFRSF1B SNORA80B CSRNP3 ITPRIP BNIP3L ATF3 ZNF331 RARA ZC3H12A CHAC1 ASTN2 VEZF1 DUSP1 SNORA80B KLF10 LPAL2 UBE2O EGR1 NR4A2 PCK1 COL7A1 PCK1 STX11 RRAGC PCK1 RBM24 GPT2 HOMER2 METRNL ARC BHLHE41 GARS C15orf41 FOS C1QTNF7 NPAS2 CSRNP3 NRCAM RGS17 VIT RGS17 UBE2O SOX9 KCTD21 NR4A3 RARA SMOX RELT GADD45B ATF3 SERTAD1 RARA BCOR BHLHE40 JUNB TINAGL1 ETS2 KLF10 GADD45B (Purple) LONRF3 COL7A1 TNFRSF1B MMP13 ATF3 SLC2A1 PIM2 CDKN1A ZC3H12A VEZF1 ARC ANKRD52 -

Genomics 93 (2009) 449–460
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Genomics 93 (2009) 449–460 Contents lists available at ScienceDirect Genomics journal homepage: www.elsevier.com/locate/ygeno Discovery of transcriptional regulators and signaling pathways in the developing pituitary gland by bioinformatic and genomic approaches Michelle L. Brinkmeier a, Shannon W. Davis a, Piero Carninci b, James W. MacDonald c, Jun Kawai b, Debashis Ghosh e,1, Yoshihide Hayashizaki b, Robert H. Lyons d, Sally A. Camper a,⁎ a Department of Human Genetics, University of Michigan Medical School, Ann Arbor, MI, 48109-5618, USA b Omics Science Center, RIKEN Yokohama Institute, Suehiro-cho, Tsurumi-ku, Yokohama, Kanagawa 230-0045, Japan c Affymetrix and cDNA Microarray Core Facility, University of Michigan Cancer Center, Ann Arbor, MI 48109-0946, USA d Department of Biological Chemistry, University of Michigan Medical School, Ann Arbor, MI, 48109-5638, USA e Department of Biostatistics, School of Public Health, University of Michigan, 48109-2029, USA article info abstract Article history: We report a catalog of the mouse embryonic pituitary gland transcriptome consisting of five cDNA libraries Received 13 December 2007 including wild type tissue from E12.5 and E14.5, Prop1df/df mutant at E14.5, and two cDNA subtractions: E14.5 Accepted 24 November 2008 WT–E14.5 Prop1df/df and E14.5 WT–E12.5 WT. DNA sequence information is assembled into a searchable Available online 11 February 2009 database with gene ontology terms representing 12,009 expressed genes. We validated coverage of the libraries by detecting most known homeobox gene transcription factor cDNAs. -

Towards Personalized Medicine in Psychiatry: Focus on Suicide
TOWARDS PERSONALIZED MEDICINE IN PSYCHIATRY: FOCUS ON SUICIDE Daniel F. Levey Submitted to the faculty of the University Graduate School in partial fulfillment of the requirements for the degree Doctor of Philosophy in the Program of Medical Neuroscience, Indiana University April 2017 ii Accepted by the Graduate Faculty, Indiana University, in partial fulfillment of the requirements for the degree of Doctor of Philosophy. Andrew J. Saykin, Psy. D. - Chair ___________________________ Alan F. Breier, M.D. Doctoral Committee Gerry S. Oxford, Ph.D. December 13, 2016 Anantha Shekhar, M.D., Ph.D. Alexander B. Niculescu III, M.D., Ph.D. iii Dedication This work is dedicated to all those who suffer, whether their pain is physical or psychological. iv Acknowledgements The work I have done over the last several years would not have been possible without the contributions of many people. I first need to thank my terrific mentor and PI, Dr. Alexander Niculescu. He has continuously given me advice and opportunities over the years even as he has suffered through my many mistakes, and I greatly appreciate his patience. The incredible passion he brings to his work every single day has been inspirational. It has been an at times painful but often exhilarating 5 years. I need to thank Helen Le-Niculescu for being a wonderful colleague and mentor. I learned a lot about organization and presentation working alongside her, and her tireless work ethic was an excellent example for a new graduate student. I had the pleasure of working with a number of great people over the years. Mikias Ayalew showed me the ropes of the lab and began my understanding of the power of algorithms. -

Global Identification of Hnrnp A1 Binding Sites for SSO-Based Splicing Modulation Gitte H
Bruun et al. BMC Biology (2016) 14:54 DOI 10.1186/s12915-016-0279-9 RESEARCH ARTICLE Open Access Global identification of hnRNP A1 binding sites for SSO-based splicing modulation Gitte H. Bruun1, Thomas K. Doktor1, Jonas Borch-Jensen1, Akio Masuda2, Adrian R. Krainer3, Kinji Ohno2 and Brage S. Andresen1* Abstract Background: Many pathogenic genetic variants have been shown to disrupt mRNA splicing. Besides splice mutations in the well-conserved splice sites, mutations in splicing regulatory elements (SREs) may deregulate splicing and cause disease. A promising therapeutic approach is to compensate for this deregulation by blocking other SREs with splice-switching oligonucleotides (SSOs). However, the location and sequence of most SREs are not well known. Results: Here, we used individual-nucleotide resolution crosslinking immunoprecipitation (iCLIP) to establish an in vivo binding map for the key splicing regulatory factor hnRNP A1 and to generate an hnRNP A1 consensus binding motif. We find that hnRNP A1 binding in proximal introns may be important for repressing exons. We show that inclusion of the alternative cassette exon 3 in SKA2 can be significantly increased by SSO-based treatment which blocks an iCLIP-identified hnRNP A1 binding site immediately downstream of the 5’ splice site. Because pseudoexons are well suited as models for constitutive exons which have been inactivated by pathogenic mutations in SREs, we used a pseudoexon in MTRR as a model and showed that an iCLIP-identified hnRNP A1 binding site downstream of the 5′ splice site can be blocked by SSOs to activate the exon. Conclusions: The hnRNP A1 binding map can be used to identify potential targets for SSO-based therapy. -

BMC Biology Biomed Central
BMC Biology BioMed Central Research article Open Access Classification and nomenclature of all human homeobox genes PeterWHHolland*†1, H Anne F Booth†1 and Elspeth A Bruford2 Address: 1Department of Zoology, University of Oxford, South Parks Road, Oxford, OX1 3PS, UK and 2HUGO Gene Nomenclature Committee, European Bioinformatics Institute (EMBL-EBI), Wellcome Trust Genome Campus, Hinxton, Cambridgeshire, CB10 1SA, UK Email: Peter WH Holland* - [email protected]; H Anne F Booth - [email protected]; Elspeth A Bruford - [email protected] * Corresponding author †Equal contributors Published: 26 October 2007 Received: 30 March 2007 Accepted: 26 October 2007 BMC Biology 2007, 5:47 doi:10.1186/1741-7007-5-47 This article is available from: http://www.biomedcentral.com/1741-7007/5/47 © 2007 Holland et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: The homeobox genes are a large and diverse group of genes, many of which play important roles in the embryonic development of animals. Increasingly, homeobox genes are being compared between genomes in an attempt to understand the evolution of animal development. Despite their importance, the full diversity of human homeobox genes has not previously been described. Results: We have identified all homeobox genes and pseudogenes in the euchromatic regions of the human genome, finding many unannotated, incorrectly annotated, unnamed, misnamed or misclassified genes and pseudogenes. -

Artin TARGETING ESTROGEN RECEPTOR AS a STRATEGY for PERSONALIZED MEDICINE in OVARIAN CANCER by Courtney Lynn Andersen A.A., Libe
TARGETING ESTROGEN RECEPTOR AS A STRATEGY FOR PERSONALIZED MEDICINE IN OVARIAN CANCER by Courtney Lynn Andersen A.A., Liberal Arts & Sciences, Middlesex Community College, 2008 B.S., Biological Sciences, University of Massachusetts Lowell, 2011 Submitted to the Graduate Faculty of the School of Medicine in partial fulfillment of the requirements for the degree of PhD in Molecular Pharmacology University of Pittsburgh artin 2016 UNIVERSITY OF PITTSBURGH SCHOOL OF MEDICINE This dissertation was presented by Courtney L. Andersen It was defended on February 16, 2016 and approved by Don DeFranco, PhD, Professor, Pharmacology & Chemical Biology Adrian Lee, PhD, Professor, Pharmacology & Chemical Biology, Anda Vlad, MD, PhD, Professor, Obstetrics, Gynecology, & Reproductive Sciences Robert Edwards, MD, Chair, Obstetrics and Gynecology, Magee-Womens Hospital of UPMC Dissertation Advisor: Steffi Oesterreich, PhD, Professor, Pharmacology & Chemical Biology ii Copyright © by Courtney L. Andersen 2016 iii TARGETING ESTROGEN RECEPTOR AS A STRATEGY FOR PERSONALIZED MEDICINE IN OVARIAN CANCER Courtney L. Andersen, PhD University of Pittsburgh, 2016 Ovarian cancer comprises a diverse set of diseases that are difficult to detect and treat successfully. Improving outcomes for ovarian cancer patients is contingent upon identifying targeted, individualized therapeutic strategies. One promising but under-utilized target is estrogen receptor-alpha (ER). ER is expressed in ~70% of epithelial ovarian cancers and epidemiologic studies implicate a role for estrogen in ovarian tumorigenesis. Further, clinical data suggest that a subset of ovarian cancer patients benefit from endocrine therapy. We hypothesized that ER drives development and progression of a subset of ovarian tumors and that outputs of ER function would identify patients who respond to endocrine therapy.