Today's Class Will Focus on the Conformations and Energies of Cycloalkanes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Conformations of Cycloalkanes
The Conformations of Cycloalkanes Ring-containing structures are a common occurrence in organic chemistry. We must, therefore spend some time studying the special characteristics of the parent cycloalkanes. Cyclical connectivity imposes constraints on the range of motion that the atoms in rings can undergo. Cyclic molecules are thus more rigid than linear or branched alkanes because cyclic structures have fewer internal degrees of freedom (that is, the motion of one atom greatly influences the motion of the others when they are connected in a ring). In this lesson we will examine structures of the common ring structures found in organic chemistry. The first four cycloalkanes are shown below. cyclopropane cyclobutane cyclopentane cyclohexane The amount of energy stored in a strained ring is estimated by comparing the experimental heat of formation to the calculated heat of formation. The calculated heat of formation is based on the notion that, in the absence of strain, each –CH2– group contributes equally to the heat of formation, in line with the behavior found for the acyclic alkanes (i.e., open chains). Thus, the calculated heat of formation varies linearly with the number of carbon atoms in the ring. Except for the 6-membered ring, the experimental values are found to have a more positive heat of formation than the calculated value owning to ring strain. The plots of calculated and experimental enthalpies of formation and their difference (i.e., ring strain) are seen in the Figure. The 3-membered ring has about 27 kcal/mol of strain. It can be seen that the 6-membered ring possesses almost no ring strain. -

Comparing Models for Measuring Ring Strain of Common Cycloalkanes
The Corinthian Volume 6 Article 4 2004 Comparing Models for Measuring Ring Strain of Common Cycloalkanes Brad A. Hobbs Georgia College Follow this and additional works at: https://kb.gcsu.edu/thecorinthian Part of the Chemistry Commons Recommended Citation Hobbs, Brad A. (2004) "Comparing Models for Measuring Ring Strain of Common Cycloalkanes," The Corinthian: Vol. 6 , Article 4. Available at: https://kb.gcsu.edu/thecorinthian/vol6/iss1/4 This Article is brought to you for free and open access by the Undergraduate Research at Knowledge Box. It has been accepted for inclusion in The Corinthian by an authorized editor of Knowledge Box. Campring Models for Measuring Ring Strain of Common Cycloalkanes Comparing Models for Measuring R..ing Strain of Common Cycloalkanes Brad A. Hobbs Dr. Kenneth C. McGill Chemistry Major Faculty Sponsor Introduction The number of carbon atoms bonded in the ring of a cycloalkane has a large effect on its energy. A molecule's energy has a vast impact on its stability. Determining the most stable form of a molecule is a usefol technique in the world of chemistry. One of the major factors that influ ence the energy (stability) of cycloalkanes is the molecule's ring strain. Ring strain is normally viewed as being directly proportional to the insta bility of a molecule. It is defined as a type of potential energy within the cyclic molecule, and is determined by the level of "strain" between the bonds of cycloalkanes. For example, propane has tl1e highest ring strain of all cycloalkanes. Each of propane's carbon atoms is sp3-hybridized. -

Cycloalkanes, Cycloalkenes, and Cycloalkynes
CYCLOALKANES, CYCLOALKENES, AND CYCLOALKYNES any important hydrocarbons, known as cycloalkanes, contain rings of carbon atoms linked together by single bonds. The simple cycloalkanes of formula (CH,), make up a particularly important homologous series in which the chemical properties change in a much more dramatic way with increasing n than do those of the acyclic hydrocarbons CH,(CH,),,-,H. The cyclo- alkanes with small rings (n = 3-6) are of special interest in exhibiting chemical properties intermediate between those of alkanes and alkenes. In this chapter we will show how this behavior can be explained in terms of angle strain and steric hindrance, concepts that have been introduced previously and will be used with increasing frequency as we proceed further. We also discuss the conformations of cycloalkanes, especially cyclo- hexane, in detail because of their importance to the chemistry of many kinds of naturally occurring organic compounds. Some attention also will be paid to polycyclic compounds, substances with more than one ring, and to cyclo- alkenes and cycloalkynes. 12-1 NOMENCLATURE AND PHYSICAL PROPERTIES OF CYCLOALKANES The IUPAC system for naming cycloalkanes and cycloalkenes was presented in some detail in Sections 3-2 and 3-3, and you may wish to review that ma- terial before proceeding further. Additional procedures are required for naming 446 12 Cycloalkanes, Cycloalkenes, and Cycloalkynes Table 12-1 Physical Properties of Alkanes and Cycloalkanes Density, Compounds Bp, "C Mp, "C diO,g ml-' propane cyclopropane butane cyclobutane pentane cyclopentane hexane cyclohexane heptane cycloheptane octane cyclooctane nonane cyclononane "At -40". bUnder pressure. polycyclic compounds, which have rings with common carbons, and these will be discussed later in this chapter. -

Are the Most Well Studied of All Ring Systems. They Have a Limited Number Of, Almost Strain Free, Conformations
Chem 201/Beauchamp Topic 6, Conformations (cyclohexanes) 1 Cyclohexanes Cyclohexane rings (six atom rings in general) are the most well studied of all ring systems. They have a limited number of, almost strain free, conformations. Because of their well defined conformational shapes, they are frequently used to study effects of orientation or steric effects when studying chemical reactions. Additionally, six atom rings are the most commonly encountered rings in nature. Cyclohexane structures do not choose to be flat. Slight twists at each carbon atom allow cyclohexane rings to assume much more comfortable conformations, which we call chair conformations. (Chairs even sound comfortable.) The chair has an up and down shape all around the ring, sort of like the zig-zag shape seen in straight chains (...time for models!). C C C C C C lounge chair - used to kick back and relax while you study your organic chemistry chair conformation Cyclohexane rings are flexible and easily allow partial rotations (twists) about the C-C single bonds. There is minimal angle strain since each carbon can approximately accommodate the 109o of the tetrahedral shape. Torsional strain energy is minimized in chair conformations since all groups are staggered relative to one another. This is easily seen in a Newman projection perspective. An added new twist to our Newman projections is a side-by-side view of parallel single bonds. If you look carefully at the structure above or use your model, you should be able to see that a parallel relationship exists for all C-C bonds across the ring from one another. -

Thermodynamic Potential of Free Energy for Thermo-Elastic-Plastic Body
Continuum Mech. Thermodyn. (2018) 30:221–232 https://doi.org/10.1007/s00161-017-0597-3 ORIGINAL ARTICLE Z. Sloderbach´ · J. Paja˛k Thermodynamic potential of free energy for thermo-elastic-plastic body Received: 22 January 2017 / Accepted: 11 September 2017 / Published online: 22 September 2017 © The Author(s) 2017. This article is an open access publication Abstract The procedure of derivation of thermodynamic potential of free energy (Helmholtz free energy) for a thermo-elastic-plastic body is presented. This procedure concerns a special thermodynamic model of a thermo-elastic-plastic body with isotropic hardening characteristics. The classical thermodynamics of irre- versible processes for material characterized by macroscopic internal parameters is used in the derivation. Thermodynamic potential of free energy may be used for practical determination of the level of stored energy accumulated in material during plastic processing applied, e.g., for industry components and other machinery parts received by plastic deformation processing. In this paper the stored energy for the simple stretching of austenitic steel will be presented. Keywords Free energy · Thermo-elastic-plastic body · Enthalpy · Exergy · Stored energy of plastic deformations · Mechanical energy dissipation · Thermodynamic reference state 1 Introduction The derivation of thermodynamic potential of free energy for a model of the thermo-elastic-plastic body with the isotropic hardening is presented. The additive form of the thermodynamic potential is assumed, see, e.g., [1–4]. Such assumption causes that the influence of plastic strains on thermo-elastic properties of the bodies is not taken into account, meaning the lack of “elastic–plastic” coupling effects, see Sloderbach´ [4–10]. -

Physical Organic Chemistry
PHYSICAL ORGANIC CHEMISTRY Yu-Tai Tao (陶雨台) Tel: (02)27898580 E-mail: [email protected] Website:http://www.sinica.edu.tw/~ytt Textbook: “Perspective on Structure and Mechanism in Organic Chemistry” by F. A. Corroll, 1998, Brooks/Cole Publishing Company References: 1. “Modern Physical Organic Chemistry” by E. V. Anslyn and D. A. Dougherty, 2005, University Science Books. Grading: One midterm (45%) one final exam (45%) and 4 quizzes (10%) homeworks Chap.1 Review of Concepts in Organic Chemistry § Quantum number and atomic orbitals Atomic orbital wavefunctions are associated with four quantum numbers: principle q. n. (n=1,2,3), azimuthal q.n. (m= 0,1,2,3 or s,p,d,f,..magnetic q. n. (for p, -1, 0, 1; for d, -2, -1, 0, 1, 2. electron spin q. n. =1/2, -1/2. § Molecular dimensions Atomic radius ionic radius, ri:size of electron cloud around an ion. covalent radius, rc:half of the distance between two atoms of same element bond to each other. van der Waal radius, rvdw:the effective size of atomic cloud around a covalently bonded atoms. - Cl Cl2 CH3Cl Bond length measures the distance between nucleus (or the local centers of electron density). Bond angle measures the angle between lines connecting different nucleus. Molecular volume and surface area can be the sum of atomic volume (or group volume) and surface area. Principle of additivity (group increment) Physical basis of additivity law: the forces between atoms in the same molecule or different molecules are very “short range”. Theoretical determination of molecular size:depending on the boundary condition. -

Klein 4A Alkanes+Isomerism
Grossman, CHE 230 4a. Alkanes. Conformational Stereoisomerism. Structural Isomerism. 4a.1 Derivatives of methane. We can replace one of the H atoms in methane with another atom or group. These atoms or groups are called heteroatoms. Examples: Methyl bromide, methyl iodide, methanol, methylamine, methanethiol. Here the central C atom is not quite as perfectly tetrahedral, but for our purposes, it is close enough. If the H atom is replaced with nothing, we have three possibilities. – • We can remove just the H nucleus and leave behind two electrons -- this gives CH3 , with sp3 hybridization. It has the same structure as ammonia, just one fewer proton (and neutron) in the nucleus in the center. + • We can take away the H nucleus and both electrons. This gives us CH3 . The hybridization here? We don't want to waste valuable low-energy s orbital in a hybrid orbital in which there are no electrons. Instead, the valuable s electron is divided equally among the six electrons (three bonds) remaining. Three bonds means three orbitals, one s and two p. That means the third p orbital remains unhybridized. This situation is called sp2 hybridization. Two p orbitals occupy a plane, so in sp2 hybridization, the three hybrid orbitals are coplanar. They point 120° apart, to the corners of a triangle. The unhybridized p orbital is perpendicular to this plane. • What if we take away the H nucleus and one electron? We have a situation in which CH3· (methyl radical) is between sp2 and sp3 hybridization. It's a shallow pyramid. These species are high in energy and don't exist for long. -

Chem 341 • Organic Chemistry I Lecture Summary 10 • September 14, 2007
Chem 341 • Organic Chemistry I Lecture Summary 10 • September 14, 2007 Chapter 4 - Stereochemistry of Alkanes and Cycloalkanes Conformations of Cycloalkanes Cyclic compounds contain something we call Ring Strain. There are three things that contribute to ring strain. Torsional strain (electron repulsion in eclipsing bonds), steric strain (atoms bumping into each other) and angle strain. Angle Strain: the strain due to bond angles being forced to expand or contract from their ideal. Sp3 hybridized atoms want to have bond angles of 109.5°. However, if the rings are very small or very large, there is no way to accommodate this angle. Thus, this increases the energy of the molecule. Heat of Combustion: the amount of heat (energy) released when a molecule burns completely with oxygen. By comparing the heat of combustion of different sized cycloalkanes, their relative energies can be obtained. The fact that the size of the ring has an influence on the total energy of the molecule indicates that there is some degree of instability associated with constraining the rings. This added energy (in addition to what would be expected from carbon and hydrogen combustion per mole) can be attributed to ring strain. n i a r t S g n i R 3 4 5 6 7 8 9 10 11 12 13 14 Ring Size Conformations of Cyclopropane Cyclopropane has a high degree of angle strain due to the highly distorted bond angles. The angle between the carbons is 60°. This cannot be accommodated by sp3-hybridized atoms. Thus, the 60° bonds actually are bent sigma bonds. -

Cycloalkanes
Cycloalkanes Adolf von Baeyer, cycloalkanes, and angle strain — The interior angles of regular polygons are given by the following formula: angle = 180(n -2)/n, where n is the number of sides to the polygon Sizes of Interior Angles in Degrees 60 90 108 120 129 135 180 The natural angle for tetrahedral carbon is 109o. Rings of 5 and 6 carbon atoms occur commonly in nature; rings of other sizes are less common. Baeyer reasoned that five and six-membered rings were common because they did not have much angle strain and therefore were more stable (contained less energy) than the other rings, which deviated from 109o by greater amounts. But great theories are sometimes brought down by experimentation. 1 Let's consider several acyclic alkanes — no rings, therefore, no strain energy. Burn each of them to form carbon dioxide and water and measure the heat of combustion. Compound DH, kcal/mole DDH, kcal/mole propane-C3H8 530.4 157.4 butane-C4H10 687.8 157.2 pentane-C5H12 845.0 157.6 hexane-C6H14 1002.6 Average 157.4 So, the average heat of combustion for a CH2 unit in an unstrained alkane is 157.4 kcal/mole. We might write: n CH2 + n(3/2) O2 -------> n CO2 + n H2O + n(157.4 kcal) Strained rings, which have more energy than unstrained ones, should give off more heat per CH2 unit when they are burned to give carbon dioxide and water. Let's see. 2 (CH2)n DH, kcal/mol DH/CH2, Strain kcal/mol energy/CH2 (CH2)3 499.8 166.6 9.2 (CH2)4 655.9 164.0 6.6 (CH2)5 793.5 158.7 1.3 (CH2)6 944.5 157.4 0 (CH2)7 1108 158.3 0.9 (CH2)8 1269 158.6 1.2 (CH2)9 1429 158.8 1.4 (CH2)10 1586 158.6 1.2 (CH2)11 1742 158.4 1.0 (CH2)12 1891 157.6 0.2 (CH2)13 2051 157.8 0.4 (CH2)14 2204 157.4 0 Bayer was right about the 3- and 4-member rings having angle strain. -

Unit-2 Conformational Isomerism
UNIT-2 CONFORMATIONAL ISOMERISM In chemistry, conformational isomerism is a form of stereoisomerism in which the isomers can be interconverted exclusively by rotations about formally single bonds. Such isomers are generally referred to as conformational isomers or conformers and specifically as rotamers when the rotation leading to different conformations is restricted (hindered) rotation, in the sense that there exists a rotational energy barrier that needs to be overcome to convert one conformer to another. Conformational isomers are thus distinct from the other classes of stereoisomers for which interconversion necessarily involves breaking and reforming of chemical bonds. The rotational barrier, or barrier to rotation, is the activation energy required to interconvert rotamers. Conformers of butane, shown in Newman projection. The two gauche as well as the anti form are staggered conformations Types of conformational isomerism Butane has three rotamers: two gauche conformers, which are enantiomeric and an anti conformer, where the four carbon centres are coplanar. The three eclipsed conformations with dihedral angles of 0°,120° and 240° are not considered to be rotamers, but are instead transition states. Some important examples of conformational isomerism include: 1. Linear alkane conformations with staggered, eclipsed and gauche conformers, and 2. Ring conformation o Cyclohexane conformations with chair and boat conformers. o Carbohydrate conformation 3. Atropisomerism- due to restricted rotation about a bond, a molecule can become chiral 4. Folding of molecules, where some shapes are stable and functional, but others are not. Conformations of Ethane While there are an infinite number of conformations about any sigma bond, in ethane two particular conformers are noteworthy and have special names. -

Alicyclic Chemistry. Lecture 2 3. Transannular Strain (Medium, 8 – 11 Membered, Rings) in Cyclooctane the Preferred Conformati
Alicyclic Chemistry. Lecture 2 3. Transannular Strain (Medium, 8 – 11 membered, rings) In cyclooctane the preferred conformation is the boat-chair, constructed from the boat and the chair conformations of two six-membered rings as follows: x boat HH chair remove bridging carbon, x On removal of the bridging carbon, x, from the bicyclo-nonane on the left, the resultant cyclooctane has two hydrogens, one either side of the ring, which point towards the space originally occupied by x. They, therefore, come within van der Waals radii of one another and this raises the energy of this conformation. Since this extra energy comes from the interaction of groups (hydrogens here) across the ring, it is known as Transannular Strain. The same is true in other medium rings as can be seen in cyclodecane, which exists in either the chair-chair conformation or the crown conformation: chair x H H remove bridging carbons x x and x H H chair H remove bond H H Crown In the former case two pairs of hydrogens get close to one another but the resultant strain is not as high as in cyclooctane, despite double the number of repulsive interactions, because cyclodecane is bigger and can flex slightly to pull the hydrogens apart. Likewise in the crown conformation some hydrogens interact across the ring. The total strain energy in rings is reflected in their heats of combustion relative to that of cyclohexane, which is generally regarded as totally strain free: The differences (∆) in heat of combustion of cycloalkanes relative to that of -1 cyclohexane in kJ mole per CH2 Ring Size (n): 3 4 5 7 8 9 10 11 12 ∆: 39 27 5.4 3.8 5.0 5.8 5.0 4.2 1.2 ∆ x n: 117 108 27 27 40 52 50 46 14 For comparison, the heats of combustion for oxiranes (114 kJ mole-1) and aziridines (113 kJ mole-1) suggest that these molecules also show the same bonding effects as cyclopropanes. -
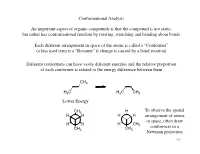
Conformational Analysis an Important Aspect of Organic Compounds Is That
Conformational Analysis An important aspect of organic compounds is that the compound is not static, but rather has conformational freedom by rotating, stretching and bending about bonds Each different arrangement in space of the atoms is called a “Conformer” (a less used term is a “Rotamer” if change is caused by a bond rotation) Different conformers can have vastly different energies and the relative proportion of each conformer is related to the energy difference between them CH3 H3C H3C CH3 Lower Energy CH3 H To observe the spatial H H H H arrangement of atoms in space, often draw H H H CH 3 conformers in a CH CH3 3 Newman projection 196 Conformational Analysis Conformers will be of different energy due to strain Sources of strain are generally categorized in one of three types: 1) Torsional strain Torsional strain is due to interactions as groups change relative position with a change in torsional bond angle Consider rotation of ethane H H Eclipsed Eclipsed is higher in H H H conformation energy than staggered H due to increased ~3 kcal/mol torsional strain y g r e n E Further rotation will H H convert eclipsed Staggered H H H H conformation back to conformation H H H H staggered H H -60˚ 0˚ 60˚ 120˚ 180˚ 240˚ 197 torsional angle Conformational Analysis 2) van der Waals strain Another source of strain is when groups are placed in positions closer than the sum of their van der Waals radii Consider rotation of butane totally eclipsed eclipsed CH H3C 3 CH H 3 H H H “totally eclipsed” CH3 H H H conformation (which has H largest groups eclipsing each other) is higher in energy than y other eclipsed conformations g r e n E “gauche” conformation is CH3 CH3 H CH3 higher in energy than anti H H (both are “staggered” H H H H H conformations) CH3 anti gauche -60˚ 0˚ 60˚ 120˚ 180˚ 240˚ 198 torsional angle Conformational Analysis The difference in energy thus affects the amount of each conformer present n-Butane CH3 H H H H Majority of CH3 compound is in the anti conformation, hardly any in CH3 H CH eclipsed 3 conformation H H Mole Fraction H Torsional Angle W.L.