Global Transcriptome Analysis of the Aphelid Paraphelidium Tribonemae Supports the Phagotrophic Origin of Fungi
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Sequence and Phylogenetic Analysis of SSU Rrna Gene of Five Microsporidia
Curr Microbiol (2010) 60:30–37 DOI 10.1007/s00284-009-9495-7 Sequence and Phylogenetic Analysis of SSU rRNA Gene of Five Microsporidia ShiNan Dong Æ ZhongYuan Shen Æ Li Xu Æ Feng Zhu Received: 21 May 2009 / Accepted: 24 August 2009 / Published online: 19 September 2009 Ó The Author(s) 2009. This article is published with open access at Springerlink.com Abstract The complete small subunit rRNA (SSU to elucidate phylogenetic relationships and taxonomic rRNA) gene sequences of five microsporidia including status of microsporidian species by analyzing information Nosema heliothidis, and four novel microsporidia isolated from SSU rRNA sequences of microsporidia. from Pieris rapae, Phyllobrotica armta, Hemerophila atrilineata, and Bombyx mori, respectively, were obtained by PCR amplification, cloning, and sequencing. Two Introduction phylogenetic trees based on SSU rRNA sequences had been constructed by using Neighbor-Joining of Phylip Microsporidia are obligate intracellular parasites that infect software and UPGMA of MEGA4.0 software. The taxo- a broad range of invertebrates and humans, they were nomic status of four novel microsporidia was determined recognized as a causative agent for many hosts including by analysis of phylogenetic relationship, length, G?C insects, fishes, rodents, and primates. In 1857, the first content, identity, and divergence of the SSU rRNA microsporidian species—N. bombycis was described by sequences. The results showed that the microsporidia iso- Nageli [1]. Until now, more than 1,200 microsporidia lated from Pieris rapae, Phyllobrotica armta, and He- species belonging to *150 genera have been reported merophila atrilineata have close phylogenetic relationship [2, 3]. with the Nosema, while another microsporidium isolated The adaptation of microsporidia to a parasitic lifestyle from Bombyx mori is closely related to the Endoreticulatus. -

The Closest Unicellular Relatives of Animals
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Current Biology, Vol. 12, 1773–1778, October 15, 2002, 2002 Elsevier Science Ltd. All rights reserved. PII S0960-9822(02)01187-9 The Closest Unicellular Relatives of Animals B.F. Lang,1,2 C. O’Kelly,1,3 T. Nerad,4 M.W. Gray,1,5 Results and Discussion and G. Burger1,2,6 1The Canadian Institute for Advanced Research The evolution of the Metazoa from single-celled protists Program in Evolutionary Biology is an issue that has intrigued biologists for more than a 2 De´ partement de Biochimie century. Early morphological and more recent ultra- Universite´ de Montre´ al structural and molecular studies have converged in sup- Succursale Centre-Ville porting the now widely accepted view that animals are Montre´ al, Que´ bec H3C 3J7 related to Fungi, choanoflagellates, and ichthyosporean Canada protists. However, controversy persists as to the spe- 3 Bigelow Laboratory for Ocean Sciences cific evolutionary relationships among these major P.O. Box 475 groups. This uncertainty is reflected in the plethora of 180 McKown Point Road published molecular phylogenies that propose virtually West Boothbay Harbor, Maine 04575 all of the possible alternative tree topologies involving 4 American Type Culture Collection Choanoflagellata, Fungi, Ichthyosporea, and Metazoa. 10801 University Boulevard For example, a monophyletic MetazoaϩChoanoflagel- Manassas, Virginia 20110 lata group has been suggested on the basis of small 5 Department of Biochemistry subunit (SSU) rDNA sequences [1, 6, 7]. Other studies and Molecular Biology using the same sequences have allied Choanoflagellata Dalhousie University with the Fungi [8], placed Choanoflagellata prior to the Halifax, Nova Scotia B3H 4H7 divergence of animals and Fungi [9], or even placed Canada them prior to the divergence of green algae and land plants [10]. -

Fungal Evolution: Major Ecological Adaptations and Evolutionary Transitions
Biol. Rev. (2019), pp. 000–000. 1 doi: 10.1111/brv.12510 Fungal evolution: major ecological adaptations and evolutionary transitions Miguel A. Naranjo-Ortiz1 and Toni Gabaldon´ 1,2,3∗ 1Department of Genomics and Bioinformatics, Centre for Genomic Regulation (CRG), The Barcelona Institute of Science and Technology, Dr. Aiguader 88, Barcelona 08003, Spain 2 Department of Experimental and Health Sciences, Universitat Pompeu Fabra (UPF), 08003 Barcelona, Spain 3ICREA, Pg. Lluís Companys 23, 08010 Barcelona, Spain ABSTRACT Fungi are a highly diverse group of heterotrophic eukaryotes characterized by the absence of phagotrophy and the presence of a chitinous cell wall. While unicellular fungi are far from rare, part of the evolutionary success of the group resides in their ability to grow indefinitely as a cylindrical multinucleated cell (hypha). Armed with these morphological traits and with an extremely high metabolical diversity, fungi have conquered numerous ecological niches and have shaped a whole world of interactions with other living organisms. Herein we survey the main evolutionary and ecological processes that have guided fungal diversity. We will first review the ecology and evolution of the zoosporic lineages and the process of terrestrialization, as one of the major evolutionary transitions in this kingdom. Several plausible scenarios have been proposed for fungal terrestralization and we here propose a new scenario, which considers icy environments as a transitory niche between water and emerged land. We then focus on exploring the main ecological relationships of Fungi with other organisms (other fungi, protozoans, animals and plants), as well as the origin of adaptations to certain specialized ecological niches within the group (lichens, black fungi and yeasts). -

Sex Is a Ubiquitous, Ancient, and Inherent Attribute of Eukaryotic Life
PAPER Sex is a ubiquitous, ancient, and inherent attribute of COLLOQUIUM eukaryotic life Dave Speijera,1, Julius Lukešb,c, and Marek Eliášd,1 aDepartment of Medical Biochemistry, Academic Medical Center, University of Amsterdam, 1105 AZ, Amsterdam, The Netherlands; bInstitute of Parasitology, Biology Centre, Czech Academy of Sciences, and Faculty of Sciences, University of South Bohemia, 370 05 Ceské Budejovice, Czech Republic; cCanadian Institute for Advanced Research, Toronto, ON, Canada M5G 1Z8; and dDepartment of Biology and Ecology, University of Ostrava, 710 00 Ostrava, Czech Republic Edited by John C. Avise, University of California, Irvine, CA, and approved April 8, 2015 (received for review February 14, 2015) Sexual reproduction and clonality in eukaryotes are mostly Sex in Eukaryotic Microorganisms: More Voyeurs Needed seen as exclusive, the latter being rather exceptional. This view Whereas absence of sex is considered as something scandalous for might be biased by focusing almost exclusively on metazoans. a zoologist, scientists studying protists, which represent the ma- We analyze and discuss reproduction in the context of extant jority of extant eukaryotic diversity (2), are much more ready to eukaryotic diversity, paying special attention to protists. We accept that a particular eukaryotic group has not shown any evi- present results of phylogenetically extended searches for ho- dence of sexual processes. Although sex is very well documented mologs of two proteins functioning in cell and nuclear fusion, in many protist groups, and members of some taxa, such as ciliates respectively (HAP2 and GEX1), providing indirect evidence for (Alveolata), diatoms (Stramenopiles), or green algae (Chlor- these processes in several eukaryotic lineages where sex has oplastida), even serve as models to study various aspects of sex- – not been observed yet. -

A Genome-Scale Phylogeny of the Kingdom Fungi
Article A genome-scale phylogeny of the kingdom Fungi Graphical Abstract Authors Yuanning Li, Jacob L. Steenwyk, Ying Chang, ..., Chris Todd Hittinger, Xing-Xing Shen, Antonis Rokas Correspondence [email protected] (X.-X.S.), [email protected] (A.R.) In Brief Li et al. analyze 290 genes from 1,644 species to infer a genome-scale phylogeny of the fungal kingdom. Analyses using different approaches and data matrices show that 85% of inferred relationships among fungi are robustly supported. The results provide a robust phylogenomic framework to explore the tempo and mode of fungal evolution. Highlights d Genome-scale phylogeny of the fungal kingdom based on 290 genes and 1,644 species d 85% of inferred phylogenetic relationships among fungi are robustly supported d Certain unresolved relationships may be due to ancient diversification events d Fungal higher rank taxonomy broadly reflects organisms’ genome sequence divergence Li et al., 2021, Current Biology 31, 1–13 April 26, 2021 ª 2021 Elsevier Inc. https://doi.org/10.1016/j.cub.2021.01.074 ll Please cite this article in press as: Li et al., A genome-scale phylogeny of the kingdom Fungi, Current Biology (2021), https://doi.org/10.1016/ j.cub.2021.01.074 ll OPEN ACCESS Article A genome-scale phylogeny of the kingdom Fungi Yuanning Li,1 Jacob L. Steenwyk,1 Ying Chang,2 Yan Wang,3,4 Timothy Y. James,5 Jason E. Stajich,3 Joseph W. Spatafora,2 Marizeth Groenewald,6 Casey W. Dunn,7 Chris Todd Hittinger,8 Xing-Xing Shen,9,* and Antonis Rokas1,10,* 1Department of Biological Sciences, -

Filling Gaps in the Microsporidian Tree: Rdna Phylogeny of Chytridiopsis Typographi (Microsporidia: Chytridiopsida)
Parasitology Research (2019) 118:169–180 https://doi.org/10.1007/s00436-018-6130-1 GENETICS, EVOLUTION, AND PHYLOGENY - ORIGINAL PAPER Filling gaps in the microsporidian tree: rDNA phylogeny of Chytridiopsis typographi (Microsporidia: Chytridiopsida) Daniele Corsaro1 & Claudia Wylezich2 & Danielle Venditti1 & Rolf Michel3 & Julia Walochnik4 & Rudolf Wegensteiner5 Received: 7 August 2018 /Accepted: 23 October 2018 /Published online: 12 November 2018 # Springer-Verlag GmbH Germany, part of Springer Nature 2018 Abstract Microsporidia are intracellular eukaryotic parasites of animals, characterized by unusual morphological and genetic features. They can be divided in three main groups, the classical microsporidians presenting all the features of the phylum and two putative primitive groups, the chytridiopsids and metchnikovellids. Microsporidia originated from microsporidia-like organisms belong- ing to a lineage of chytrid-like endoparasites basal or sister to the Fungi. Genetic and genomic data are available for all members, except chytridiopsids. Herein, we filled this gap by obtaining the rDNA sequence (SSU-ITS-partial LSU) of Chytridiopsis typographi (Chytridiopsida), a parasite of bark beetles. Our rDNA molecular phylogenies indicate that Chytridiopsis branches earlier than metchnikovellids, commonly thought ancestral, forming the more basal lineage of the Microsporidia. Furthermore, our structural analyses showed that only classical microsporidians present 16S-like SSU rRNA and 5.8S/LSU rRNA gene fusion, whereas the standard eukaryote rRNA gene structure, although slightly reduced, is still preserved in the primitive microsporidians, including 18S-like SSU rRNA with conserved core helices, and ITS2-like separating 5.8S from LSU. Overall, our results are consistent with the scenario of an evolution from microsporidia-like rozellids to microsporidians, however suggesting for metchnikovellids a derived position, probably related to marine transition and adaptation to hyperparasitism. -

Multigene Eukaryote Phylogeny Reveals the Likely Protozoan Ancestors of Opis- Thokonts (Animals, Fungi, Choanozoans) and Amoebozoa
Accepted Manuscript Multigene eukaryote phylogeny reveals the likely protozoan ancestors of opis- thokonts (animals, fungi, choanozoans) and Amoebozoa Thomas Cavalier-Smith, Ema E. Chao, Elizabeth A. Snell, Cédric Berney, Anna Maria Fiore-Donno, Rhodri Lewis PII: S1055-7903(14)00279-6 DOI: http://dx.doi.org/10.1016/j.ympev.2014.08.012 Reference: YMPEV 4996 To appear in: Molecular Phylogenetics and Evolution Received Date: 24 January 2014 Revised Date: 2 August 2014 Accepted Date: 11 August 2014 Please cite this article as: Cavalier-Smith, T., Chao, E.E., Snell, E.A., Berney, C., Fiore-Donno, A.M., Lewis, R., Multigene eukaryote phylogeny reveals the likely protozoan ancestors of opisthokonts (animals, fungi, choanozoans) and Amoebozoa, Molecular Phylogenetics and Evolution (2014), doi: http://dx.doi.org/10.1016/ j.ympev.2014.08.012 This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. 1 1 Multigene eukaryote phylogeny reveals the likely protozoan ancestors of opisthokonts 2 (animals, fungi, choanozoans) and Amoebozoa 3 4 Thomas Cavalier-Smith1, Ema E. Chao1, Elizabeth A. Snell1, Cédric Berney1,2, Anna Maria 5 Fiore-Donno1,3, and Rhodri Lewis1 6 7 1Department of Zoology, University of Oxford, South Parks Road, Oxford OX1 3PS, UK. -
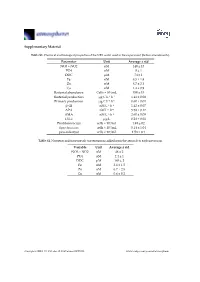
Supplementary Material Parameter Unit Average ± Std NO3 + NO2 Nm
Supplementary Material Table S1. Chemical and biological properties of the NRS water used in the experiment (before amendments). Parameter Unit Average ± std NO3 + NO2 nM 140 ± 13 PO4 nM 8 ± 1 DOC μM 74 ± 1 Fe nM 8.5 ± 1.8 Zn nM 8.7 ± 2.1 Cu nM 1.4 ± 0.9 Bacterial abundance Cells × 104/mL 350 ± 15 Bacterial production μg C L−1 h−1 1.41 ± 0.08 Primary production μg C L−1 h−1 0.60 ± 0.01 β-Gl nM L−1 h−1 1.42 ± 0.07 APA nM L−1 h−1 5.58 ± 0.17 AMA nM L−1·h−1 2.60 ± 0.09 Chl-a μg/L 0.28 ± 0.01 Prochlorococcus cells × 104/mL 1.49 ± 02 Synechococcus cells × 104/mL 5.14 ± 1.04 pico-eukaryot cells × 103/mL 1.58 × 0.1 Table S2. Nutrients and trace metals concentrations added from the aerosols to each mesocosm. Variable Unit Average ± std NO3 + NO2 nM 48 ± 2 PO4 nM 2.4 ± 1 DOC μM 165 ± 2 Fe nM 2.6 ± 1.5 Zn nM 6.7 ± 2.5 Cu nM 0.6 ± 0.2 Atmosphere 2019, 10, 358; doi:10.3390/atmos10070358 www.mdpi.com/journal/atmosphere Atmosphere 2019, 10, 358 2 of 6 Table S3. ANOVA test results between control, ‘UV-treated’ and ‘live-dust’ treatments at 20 h or 44 h, with significantly different values shown in bold. ANOVA df Sum Sq Mean Sq F Value p-value Chl-a 20 H 2, 6 0.03, 0.02 0.02, 0 4.52 0.0634 44 H 2, 6 0.02, 0 0.01, 0 23.13 0.002 Synechococcus Abundance 20 H 2, 7 8.23 × 107, 4.11 × 107 4.11 × 107, 4.51 × 107 0.91 0.4509 44 H 2, 7 5.31 × 108, 6.97 × 107 2.65 × 108, 1.16 × 107 22.84 0.0016 Prochlorococcus Abundance 20 H 2, 8 4.22 × 107, 2.11 × 107 2.11 × 107, 2.71 × 106 7.77 0.0216 44 H 2, 8 9.02 × 107, 1.47 × 107 4.51 × 107, 2.45 × 106 18.38 0.0028 Pico-eukaryote -

Predatory Flagellates – the New Recently Discovered Deep Branches of the Eukaryotic Tree and Their Evolutionary and Ecological Significance
Protistology 14 (1), 15–22 (2020) Protistology Predatory flagellates – the new recently discovered deep branches of the eukaryotic tree and their evolutionary and ecological significance Denis V. Tikhonenkov Papanin Institute for Biology of Inland Waters, Russian Academy of Sciences, Borok, 152742, Russia | Submitted March 20, 2020 | Accepted April 6, 2020 | Summary Predatory protists are poorly studied, although they are often representing important deep-branching evolutionary lineages and new eukaryotic supergroups. This short review/opinion paper is inspired by the recent discoveries of various predatory flagellates, which form sister groups of the giant eukaryotic clusters on phylogenetic trees, and illustrate an ancestral state of one or another supergroup of eukaryotes. Here we discuss their evolutionary and ecological relevance and show that the study of such protists may be essential in addressing previously puzzling evolutionary problems, such as the origin of multicellular animals, the plastid spread trajectory, origins of photosynthesis and parasitism, evolution of mitochondrial genomes. Key words: evolution of eukaryotes, heterotrophic flagellates, mitochondrial genome, origin of animals, photosynthesis, predatory protists, tree of life Predatory flagellates and diversity of eu- of the hidden diversity of protists (Moon-van der karyotes Staay et al., 2000; López-García et al., 2001; Edg- comb et al., 2002; Massana et al., 2004; Richards The well-studied multicellular animals, plants and Bass, 2005; Tarbe et al., 2011; de Vargas et al., and fungi immediately come to mind when we hear 2015). In particular, several prevailing and very abun- the term “eukaryotes”. However, these groups of dant ribogroups such as MALV, MAST, MAOP, organisms represent a minority in the real diversity MAFO (marine alveolates, stramenopiles, opistho- of evolutionary lineages of eukaryotes. -

Protist Phylogeny and the High-Level Classification of Protozoa
Europ. J. Protistol. 39, 338–348 (2003) © Urban & Fischer Verlag http://www.urbanfischer.de/journals/ejp Protist phylogeny and the high-level classification of Protozoa Thomas Cavalier-Smith Department of Zoology, University of Oxford, South Parks Road, Oxford, OX1 3PS, UK; E-mail: [email protected] Received 1 September 2003; 29 September 2003. Accepted: 29 September 2003 Protist large-scale phylogeny is briefly reviewed and a revised higher classification of the kingdom Pro- tozoa into 11 phyla presented. Complementary gene fusions reveal a fundamental bifurcation among eu- karyotes between two major clades: the ancestrally uniciliate (often unicentriolar) unikonts and the an- cestrally biciliate bikonts, which undergo ciliary transformation by converting a younger anterior cilium into a dissimilar older posterior cilium. Unikonts comprise the ancestrally unikont protozoan phylum Amoebozoa and the opisthokonts (kingdom Animalia, phylum Choanozoa, their sisters or ancestors; and kingdom Fungi). They share a derived triple-gene fusion, absent from bikonts. Bikonts contrastingly share a derived gene fusion between dihydrofolate reductase and thymidylate synthase and include plants and all other protists, comprising the protozoan infrakingdoms Rhizaria [phyla Cercozoa and Re- taria (Radiozoa, Foraminifera)] and Excavata (phyla Loukozoa, Metamonada, Euglenozoa, Percolozoa), plus the kingdom Plantae [Viridaeplantae, Rhodophyta (sisters); Glaucophyta], the chromalveolate clade, and the protozoan phylum Apusozoa (Thecomonadea, Diphylleida). Chromalveolates comprise kingdom Chromista (Cryptista, Heterokonta, Haptophyta) and the protozoan infrakingdom Alveolata [phyla Cilio- phora and Miozoa (= Protalveolata, Dinozoa, Apicomplexa)], which diverged from a common ancestor that enslaved a red alga and evolved novel plastid protein-targeting machinery via the host rough ER and the enslaved algal plasma membrane (periplastid membrane). -

The Evolution of Multicellularity and Early Animal Genomes Nina M Brooke and Peter WH Hollandã
599 The evolution of multicellularity and early animal genomes Nina M Brooke and Peter WH Hollandà Several independent molecular datasets, including complete although some expressed sequence tag screens have mtDNA sequence, indicate that Choanozoa are most closely commenced (Hydra EST Database: http://hampson- related to multicellular animals. There is still confusion pc2.ics.uci.edu/blast/jf/; [1,2]) and gene-expression data concerning basal animal phylogeny, although recent data are accumulating (for cnidarians, these are being indicate that Placozoa are not degenerate cnidarians and hence assembled into a web-accessible database [3]). (along with sponges) occupy a pivotal position. The transition in evolution from diploblast to bilaterian animals is becoming better Models and mechanisms understood, with gene expression data arguing that cnidarians Some organisational principles are shared by different have forerunners of the anteroposterior and dorsoventral body groups of multicellular organisms, such as division of axes, and even a putative homologue of mesoderm. The labour, differentiation and cell communication. Wolpert homeobox and kinase gene families have been further analysed and Szathmary [4] propose that development from a single in basal animals, although more data are required to enable cell is an additional feature shared by all those multicellular detailed comparison with Bilateria. organisms that evolved complexity and diversity. Their reasoning is that an egg creates a genetic bottleneck in Addresses development, removing the potential for conflict between Department of Zoology, University of Oxford, South Parks Road, somatic cells, and allowing coordination to evolve. Oxford OX1 3PS, UK Ãe-mail: [email protected] When we consider molecular mechanisms responsible for that coordination, however, there is no reason to Current Opinion in Genetics & Development 2003, 13:599–603 suspect there will be much similarity between different multicellular lineages. -

Protistology Molecular Phylogeny of Aphelidium Arduennense Sp. Nov
Protistology 13 (4), 192–198 (2019) Protistology Molecular phylogeny of Aphelidium arduennense sp. nov. – new representative of Aphelida (Opis- thosporidia) Victoria S. Tcvetkova1, Natalia A. Zorina1, Maria A. Mamkaeva1 and Sergey A. Karpov1,2 1 St. Petersburg State University, St. Petersburg 199034, Russia 2 Zoological Institute, Russian Academy of Sciences, St. Petersburg 199034, Russia | Submitted November 14, 2019 | Accepted December 2, 2019 | Summary Aphelids (Aphelida) are poorly known parasitoids of algae that have raised considerable interest because of their phylogenetic position as phagotrophic protists sister to Fungi. Together with Rozellida and Microsporidia they have been classified in the Opisthosporidia but seem to be more closely related to the Fungi rather than to the Cryptomycota and Microsporidia, the other members of the Opisthosporidia. Molecular environmental studies have revealed high genetic diversity within the aphelids, but only four genera have been described: Aphelidium, Amoeboaphelidium, Paraphelidium and Pseudaphelidium. Here, we describe the life cycle of a new species of Aphelidium, Aph. arduennense. Molecular phylogenetic analysis of its 18S rRNA indicates that Aph. arduennense is sister to Aph. tribonematis, and together with Aph. melosirae they form a monophyletic cluster. Within the aphelids, this cluster is distantly related to Paraphelidium and Amoeboaphelidium. Key words: aphelids, Holomycota, Opisthosporidia, Rozellosporidia, taxonomy Introduction Rozellosporidia (Cryptomycota) formed the super- phylum Opisthosporidia, the deepest branch Aphelids are a divergent group of intracellular of the Holomycota lineage, separated from the parasitoids of green, yellow-green and diatom algae Fungi (Karpov et al., 2014a; Letcher et al., 2015; (Gromov, 2000; Karpov et al., 2014a). The four 2017; Torruella et al., 2015). Several biological known genera have different ecological preferences: peculiarities of the aphelids do not conform the Aphelidium, Amoeboaphelidium and Paraphelidium classical definition of the Fungi.