The Genome Sequence of the Octocoral Paramuricea Clavata – a Key Resource to Study the Impact of Climate Change in the Mediterranean
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Octocoral Diversity of Balıkçı Island, the Marmara Sea
J. Black Sea/Mediterranean Environment Vol. 19, No. 1: 46-57 (2013) RESEARCH ARTICLE Octocoral diversity of Balıkçı Island, the Marmara Sea Eda Nur Topçu1,2*, Bayram Öztürk1,2 1Faculty of Fisheries, Istanbul University, Ordu St., No. 200, 34470, Laleli, Istanbul, TURKEY 2Turkish Marine Research Foundation (TUDAV), P.O. Box: 10, Beykoz, Istanbul, TURKEY *Corresponding author: [email protected] Abstract We investigated the octocoral diversity of Balıkçı Island in the Marmara Sea. Three sites were sampled by diving, from 20 to 45 m deep. Nine species were found, two of which are first records for Turkish fauna: Alcyonium coralloides and Paralcyonium spinulosum. Scientific identification of Alcyonium acaule in the Turkish seas was also done for the first time in this study. Key words: Octocoral, soft coral, gorgonian, diversity, Marmara Sea. Introduction The Marmara Sea is a semi-enclosed sea connecting the Black Sea to the Aegean Sea via the Turkish Straits System, with peculiar oceanographic, ecological and geomorphologic characteristics (Öztürk and Öztürk 1996). The benthic fauna consists of Black Sea species until approximately 20 meters around the Prince Islands area, where Mediterranean species take over due to the two layer stratification in the Marmara Sea. The Sea of Marmara, together with the straits of Istanbul and Çanakkale, serves as an ecological barrier, a biological corridor and an acclimatization zone for the biota of Mediterranean and Black Seas (Öztürk and Öztürk 1996). The Islands in the Sea of Marmara constitute habitats particularly for hard bottom communities of Mediterranean origin. Ten Octocoral species were reported by Demir (1954) from the Marmara Sea but amongst them, Gorgonia flabellum was probably reported by mistake and 46 should not be considered as a valid record from the Sea of Marmara. -

Di Camillo Et Al 2017
This is a post-peer-review, pre-copyedit version of an article published in Biodiversity and Conservation on 23 December 2017 (First Online). The final authenticated version is available online at: https://doi.org/10.1007/s10531-017-1492-8 https://link.springer.com/article/10.1007%2Fs10531-017-1492-8 An embargo period of 12 months applies to this Journal. This paper has received funding from the European Union (EU)’s H2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No 643712 to the project Green Bubbles RISE for sustainable diving (Green Bubbles). This paper reflects only the authors’ view. The Research Executive Agency is not responsible for any use that may be made of the information it contains. © 2017. This manuscript version is made available under the CC-BY-NC-ND 4.0 AUTHORS' ACCEPTED MANUSCRIPT Building a baseline for habitat-forming corals by a multi-source approach, including Web Ecological Knowledge - Cristina G Di Camillo, Department of Life and Environmental Sciences, Marche Polytechnic University, CoNISMa, Ancona, Italy, [email protected] - Massimo Ponti, Department of Biological, Geological and Environmental Sciences and Interdepartmental Research Centre for Environmental SciencesUniversity of Bologna, CoNISMa, Ravenna, Italy - Giorgio Bavestrello, Department of Earth, Environment and Life Sciences, University of Genoa, CoNISMa, Genoa, Italy - Maja Krzelj, Department of Marine Studies, University of Split, Split, Croatia - Carlo Cerrano, Department of Life and Environmental Sciences, Marche Polytechnic University, CoNISMa, Ancona, Italy Received: 12 January 2017 Revised: 10 December 2017 Accepted: 14 December 2017 First online: 23 December 2017 Cite as: Di Camillo, C.G., Ponti, M., Bavestrello, G. -

A Family of GFP-Like Proteins with Different Spectral Properties In
Biology Direct BioMed Central Research Open Access A family of GFP-like proteins with different spectral properties in lancelet Branchiostoma floridae Diana Baumann1, Malcolm Cook1, Limei Ma1, Arcady Mushegian*1,2, Erik Sanders1, Joel Schwartz1 and C Ron Yu1,3 Address: 1Stowers Institute for Medical Research, 1000 E 50th St., Kansas City, MO, 64110, USA, 2Department of Microbiology, Molecular Genetics, and Immunology, University of Kansas, Kansas City, KS, 66160, USA and 3Department of Anatomy and Cell Biology, University of Kansas Medical Center, Kansas City, KS, 66160, USA Email: Diana Baumann - [email protected]; Malcolm Cook - [email protected]; Limei Ma - [email protected]; Arcady Mushegian* - [email protected]; Erik Sanders - [email protected]; Joel Schwartz - [email protected]; C Ron Yu - [email protected] * Corresponding author Published: 3 July 2008 Received: 11 June 2008 Accepted: 3 July 2008 Biology Direct 2008, 3:28 doi:10.1186/1745-6150-3-28 This article is available from: http://www.biology-direct.com/content/3/1/28 © 2008 Baumann et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Members of the green fluorescent protein (GFP) family share sequence similarity and the 11-stranded β-barrel fold. Fluorescence or bright coloration, observed in many members of this family, is enabled by the intrinsic properties of the polypeptide chain itself, without the requirement for cofactors. -

Coelenterata: Anthozoa), with Diagnoses of New Taxa
PROC. BIOL. SOC. WASH. 94(3), 1981, pp. 902-947 KEY TO THE GENERA OF OCTOCORALLIA EXCLUSIVE OF PENNATULACEA (COELENTERATA: ANTHOZOA), WITH DIAGNOSES OF NEW TAXA Frederick M. Bayer Abstract.—A serial key to the genera of Octocorallia exclusive of the Pennatulacea is presented. New taxa introduced are Olindagorgia, new genus for Pseudopterogorgia marcgravii Bayer; Nicaule, new genus for N. crucifera, new species; and Lytreia, new genus for Thesea plana Deich- mann. Ideogorgia is proposed as a replacement ñame for Dendrogorgia Simpson, 1910, not Duchassaing, 1870, and Helicogorgia for Hicksonella Simpson, December 1910, not Nutting, May 1910. A revised classification is provided. Introduction The key presented here was an essential outgrowth of work on a general revisión of the octocoral fauna of the western part of the Atlantic Ocean. The far-reaching zoogeographical affinities of this fauna made it impossible in the course of this study to ignore genera from any part of the world, and it soon became clear that many of them require redefinition according to modern taxonomic standards. Therefore, the type-species of as many genera as possible have been examined, often on the basis of original type material, and a fully illustrated generic revisión is in course of preparation as an essential first stage in the redescription of western Atlantic species. The key prepared to accompany this generic review has now reached a stage that would benefit from a broader and more objective testing under practical conditions than is possible in one laboratory. For this reason, and in order to make the results of this long-term study available, even in provisional form, not only to specialists but also to the growing number of ecologists, biochemists, and physiologists interested in octocorals, the key is now pre- sented in condensed form with minimal illustration. -

Updated Chronology of Mass Mortality Events Hitting Gorgonians in the Western Mediterranean Sea (Modified and Updated from Calvo Et Al
The following supplement accompanies the article Mass mortality hits gorgonian forests at Montecristo Island Eva Turicchia*, Marco Abbiati, Michael Sweet and Massimo Ponti *Corresponding author: [email protected] Diseases of Aquatic Organisms 131: 79–85 (2018) Table S1. Updated chronology of mass mortality events hitting gorgonians in the western Mediterranean Sea (modified and updated from Calvo et al. 2011). Year Locations Scale Depth range Species References (m) 1983 La Ciotat (Ligurian Sea) Local 0 to 20 Eunicella singularis Harmelin 1984 Corallium rubrum 1986 Portofino Promontory (Ligurian Local 0 to 20 Eunicella cavolini Bavestrello & Boero 1988 Sea) 1989 Montecristo Island (Tyrrhenian Local - Paramuricea clavata Guldenschuh in Bavestrello et al. 1994 Sea) 1992 Medes Islands (north-western Local 0 to 14 Paramuricea clavata Coma & Zabala 1992 Mediterranean Sea), Port-Cros 10 to 45 Harmelin & Marinopoulos 1994 National Park 1993 Strait of Messina (Tyrrhenian Local 20 to 39 Paramuricea clavata Mistri & Ceccherelli 1996 Sea), Portofino Promontory Bavestrello et al. 1994 (Ligurian Sea) 1999 Coast of Provence and Ligurian Regional 0 to 45 Paramuricea clavata Cerrano et al. 2000 Sea, Balearic Islands (north- Eunicella singularis Perez et al. 2000 western Mediterranean Sea), Eunicella cavolini Garrabou et al. 2001 Gulf of La Spezia, Port-Cros Eunicella verrucosa Linares et al. 2005 National Park, coast of Calafuria Corallium rubrum Bramanti et al. 2005 (Tyrrhenian Sea) Leptogorgia Coma et al. 2006 sarmentosa Cupido et al. 2008 Crisci et al. 2011 2001 Tavolara Island (Tyrrhenian Sea) Local 10 to 45 Paramuricea Calvisi et al. 2003 clavata Eunicella cavolini 2002 Ischia and Procida Islands Local 15 to 20 Paramuricea clavata Gambi et al. -

Microbiomes of Gall-Inducing Copepod Crustaceans from the Corals Stylophora Pistillata (Scleractinia) and Gorgonia Ventalina
www.nature.com/scientificreports OPEN Microbiomes of gall-inducing copepod crustaceans from the corals Stylophora pistillata Received: 26 February 2018 Accepted: 18 July 2018 (Scleractinia) and Gorgonia Published: xx xx xxxx ventalina (Alcyonacea) Pavel V. Shelyakin1,2, Sofya K. Garushyants1,3, Mikhail A. Nikitin4, Sofya V. Mudrova5, Michael Berumen 5, Arjen G. C. L. Speksnijder6, Bert W. Hoeksema6, Diego Fontaneto7, Mikhail S. Gelfand1,3,4,8 & Viatcheslav N. Ivanenko 6,9 Corals harbor complex and diverse microbial communities that strongly impact host ftness and resistance to diseases, but these microbes themselves can be infuenced by stresses, like those caused by the presence of macroscopic symbionts. In addition to directly infuencing the host, symbionts may transmit pathogenic microbial communities. We analyzed two coral gall-forming copepod systems by using 16S rRNA gene metagenomic sequencing: (1) the sea fan Gorgonia ventalina with copepods of the genus Sphaerippe from the Caribbean and (2) the scleractinian coral Stylophora pistillata with copepods of the genus Spaniomolgus from the Saudi Arabian part of the Red Sea. We show that bacterial communities in these two systems were substantially diferent with Actinobacteria, Alphaproteobacteria, and Betaproteobacteria more prevalent in samples from Gorgonia ventalina, and Gammaproteobacteria in Stylophora pistillata. In Stylophora pistillata, normal coral microbiomes were enriched with the common coral symbiont Endozoicomonas and some unclassifed bacteria, while copepod and gall-tissue microbiomes were highly enriched with the family ME2 (Oceanospirillales) or Rhodobacteraceae. In Gorgonia ventalina, no bacterial group had signifcantly diferent prevalence in the normal coral tissues, copepods, and injured tissues. The total microbiome composition of polyps injured by copepods was diferent. -
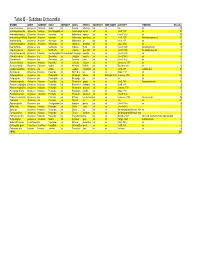
Table B – Subclass Octocorallia
Table B – Subclass Octocorallia BINOMEN ORDER SUBORDER FAMILY SUBFAMILY GENUS SPECIES SUBSPECIES COMN_NAMES AUTHORITY SYNONYMS #Records Acanella arbuscula Alcyonacea Calcaxonia Isididae n/a Acanella arbuscula n/a n/a n/a n/a 59 Acanthogorgia armata Alcyonacea Holaxonia Acanthogorgiidae n/a Acanthogorgia armata n/a n/a Verrill, 1878 n/a 95 Anthomastus agassizii Alcyonacea Alcyoniina Alcyoniidae n/a Anthomastus agassizii n/a n/a (Verrill, 1922) n/a 35 Anthomastus grandiflorus Alcyonacea Alcyoniina Alcyoniidae n/a Anthomastus grandiflorus n/a n/a Verrill, 1878 Anthomastus purpureus 37 Anthomastus sp. Alcyonacea Alcyoniina Alcyoniidae n/a Anthomastus sp. n/a n/a Verrill, 1878 n/a 1 Anthothela grandiflora Alcyonacea Scleraxonia Anthothelidae n/a Anthothela grandiflora n/a n/a (Sars, 1856) n/a 24 Capnella florida Alcyonacea n/a Nephtheidae n/a Capnella florida n/a n/a (Verrill, 1869) Eunephthya florida 44 Capnella glomerata Alcyonacea n/a Nephtheidae n/a Capnella glomerata n/a n/a (Verrill, 1869) Eunephthya glomerata 4 Chrysogorgia agassizii Alcyonacea Holaxonia Acanthogorgiidae Chrysogorgiidae Chrysogorgia agassizii n/a n/a (Verrill, 1883) n/a 2 Clavularia modesta Alcyonacea n/a Clavulariidae n/a Clavularia modesta n/a n/a (Verrill, 1987) n/a 6 Clavularia rudis Alcyonacea n/a Clavulariidae n/a Clavularia rudis n/a n/a (Verrill, 1922) n/a 1 Gersemia fruticosa Alcyonacea Alcyoniina Alcyoniidae n/a Gersemia fruticosa n/a n/a Marenzeller, 1877 n/a 3 Keratoisis flexibilis Alcyonacea Calcaxonia Isididae n/a Keratoisis flexibilis n/a n/a Pourtales, 1868 n/a 1 Lepidisis caryophyllia Alcyonacea n/a Isididae n/a Lepidisis caryophyllia n/a n/a Verrill, 1883 Lepidisis vitrea 13 Muriceides sp. -

Comprehensive Phylogenomic Analyses Resolve Cnidarian Relationships and the Origins of Key Organismal Traits
Comprehensive phylogenomic analyses resolve cnidarian relationships and the origins of key organismal traits Ehsan Kayal1,2, Bastian Bentlage1,3, M. Sabrina Pankey5, Aki H. Ohdera4, Monica Medina4, David C. Plachetzki5*, Allen G. Collins1,6, Joseph F. Ryan7,8* Authors Institutions: 1. Department of Invertebrate Zoology, National Museum of Natural History, Smithsonian Institution 2. UPMC, CNRS, FR2424, ABiMS, Station Biologique, 29680 Roscoff, France 3. Marine Laboratory, university of Guam, UOG Station, Mangilao, GU 96923, USA 4. Department of Biology, Pennsylvania State University, University Park, PA, USA 5. Department of Molecular, Cellular and Biomedical Sciences, University of New Hampshire, Durham, NH, USA 6. National Systematics Laboratory, NOAA Fisheries, National Museum of Natural History, Smithsonian Institution 7. Whitney Laboratory for Marine Bioscience, University of Florida, St Augustine, FL, USA 8. Department of Biology, University of Florida, Gainesville, FL, USA PeerJ Preprints | https://doi.org/10.7287/peerj.preprints.3172v1 | CC BY 4.0 Open Access | rec: 21 Aug 2017, publ: 21 Aug 20171 Abstract Background: The phylogeny of Cnidaria has been a source of debate for decades, during which nearly all-possible relationships among the major lineages have been proposed. The ecological success of Cnidaria is predicated on several fascinating organismal innovations including symbiosis, colonial body plans and elaborate life histories, however, understanding the origins and subsequent diversification of these traits remains difficult due to persistent uncertainty surrounding the evolutionary relationships within Cnidaria. While recent phylogenomic studies have advanced our knowledge of the cnidarian tree of life, no analysis to date has included genome scale data for each major cnidarian lineage. Results: Here we describe a well-supported hypothesis for cnidarian phylogeny based on phylogenomic analyses of new and existing genome scale data that includes representatives of all cnidarian classes. -

Differences in Aquatic Versus Terrestrial Life History Strategies
1 Longevity, body dimension and reproductive mode drive 2 differences in aquatic versus terrestrial life history strategies 3 A manuscript for consideration as Research Article in Functional Ecology 4 5 Running title: Life history strategies across environments 6 Word count: abstract (222); main text (5624); References (69). 7 Figures: 4; Tables: 2; Online Materials: 2 1 8 Abstract 9 1. Aquatic and terrestrial environments display stark differences in key 10 environmental factors and phylogenetic composition but their 11 consequences for the evolution of species’ life history strategies remain 12 poorly understood. 13 2. Here, we examine whether and how life history strategies vary between 14 terrestrial and aquatic species. We use demographic information for 685 15 terrestrial and 122 aquatic animal and plant species to estimate key life 16 history traits. We then use phylogenetically corrected least squares 17 regression to explore potential differences in trade-offs between life history 18 traits between both environments. We contrast life history strategies of 19 aquatic vs. terrestrial species in a principal component analysis while 20 accounting for body dimensions and phylogenetic relationships. 21 3. Our results show that the same trade-offs structure terrestrial and aquatic 22 life histories, resulting in two dominant axes of variation that describe 23 species’ pace-of-life and reproductive strategies. Terrestrial plants display 24 a large diversity of strategies, including the longest-lived species in this 25 study. Aquatic animals exhibit higher reproductive frequency than 26 terrestrial animals. When correcting for body size, mobile and sessile 27 terrestrial organisms show slower paces of life than aquatic ones. -
![Genetic Divergence and Polyphyly in the Octocoral Genus Swiftia [Cnidaria: Octocorallia], Including a Species Impacted by the DWH Oil Spill](https://docslib.b-cdn.net/cover/9917/genetic-divergence-and-polyphyly-in-the-octocoral-genus-swiftia-cnidaria-octocorallia-including-a-species-impacted-by-the-dwh-oil-spill-739917.webp)
Genetic Divergence and Polyphyly in the Octocoral Genus Swiftia [Cnidaria: Octocorallia], Including a Species Impacted by the DWH Oil Spill
diversity Article Genetic Divergence and Polyphyly in the Octocoral Genus Swiftia [Cnidaria: Octocorallia], Including a Species Impacted by the DWH Oil Spill Janessy Frometa 1,2,* , Peter J. Etnoyer 2, Andrea M. Quattrini 3, Santiago Herrera 4 and Thomas W. Greig 2 1 CSS Dynamac, Inc., 10301 Democracy Lane, Suite 300, Fairfax, VA 22030, USA 2 Hollings Marine Laboratory, NOAA National Centers for Coastal Ocean Sciences, National Ocean Service, National Oceanic and Atmospheric Administration, 331 Fort Johnson Rd, Charleston, SC 29412, USA; [email protected] (P.J.E.); [email protected] (T.W.G.) 3 Department of Invertebrate Zoology, National Museum of Natural History, Smithsonian Institution, 10th and Constitution Ave NW, Washington, DC 20560, USA; [email protected] 4 Department of Biological Sciences, Lehigh University, 111 Research Dr, Bethlehem, PA 18015, USA; [email protected] * Correspondence: [email protected] Abstract: Mesophotic coral ecosystems (MCEs) are recognized around the world as diverse and ecologically important habitats. In the northern Gulf of Mexico (GoMx), MCEs are rocky reefs with abundant black corals and octocorals, including the species Swiftia exserta. Surveys following the Deepwater Horizon (DWH) oil spill in 2010 revealed significant injury to these and other species, the restoration of which requires an in-depth understanding of the biology, ecology, and genetic diversity of each species. To support a larger population connectivity study of impacted octocorals in the Citation: Frometa, J.; Etnoyer, P.J.; GoMx, this study combined sequences of mtMutS and nuclear 28S rDNA to confirm the identity Quattrini, A.M.; Herrera, S.; Greig, Swiftia T.W. -

Vulnerable Forests of the Pink Sea Fan Eunicella Verrucosa in the Mediterranean Sea
diversity Article Vulnerable Forests of the Pink Sea Fan Eunicella verrucosa in the Mediterranean Sea Giovanni Chimienti 1,2 1 Dipartimento di Biologia, Università degli Studi di Bari, Via Orabona 4, 70125 Bari, Italy; [email protected]; Tel.: +39-080-544-3344 2 CoNISMa, Piazzale Flaminio 9, 00197 Roma, Italy Received: 14 April 2020; Accepted: 28 April 2020; Published: 30 April 2020 Abstract: The pink sea fan Eunicella verrucosa (Cnidaria, Anthozoa, Alcyonacea) can form coral forests at mesophotic depths in the Mediterranean Sea. Despite the recognized importance of these habitats, they have been scantly studied and their distribution is mostly unknown. This study reports the new finding of E. verrucosa forests in the Mediterranean Sea, and the updated distribution of this species that has been considered rare in the basin. In particular, one site off Sanremo (Ligurian Sea) was characterized by a monospecific population of E. verrucosa with 2.3 0.2 colonies m 2. By combining ± − new records, literature, and citizen science data, the species is believed to be widespread in the basin with few or isolated colonies, and 19 E. verrucosa forests were identified. The overall associated community showed how these coral forests are essential for species of conservation interest, as well as for species of high commercial value. For this reason, proper protection and management strategies are necessary. Keywords: Anthozoa; Alcyonacea; gorgonian; coral habitat; coral forest; VME; biodiversity; mesophotic; citizen science; distribution 1. Introduction Arborescent corals such as antipatharians and alcyonaceans can form mono- or multispecific animal forests that represent vulnerable marine ecosystems of great ecological importance [1–4]. -

Curriculum Vitae
CURRICULUM VITAE Elizabeth (Beth) Anne Horvath Education: --May, 1976, Bachelor of Arts, Biology--Westmont College --August, 1981, Masters of Science, Biology--Cal. State Univ., Long Beach Dissertation Title: Experiences in Marine Science, an Introductory Text. Supplemental Research Projects: Pelagic Snails; Development in Pelagic Tunicates; SEM studies of Luminescence in Pelagic Tunicates --Selected Graduate Level Courses beyond Master's; subjects include-- Channel Island Biology, Deep Sea Biology, Conservation Biology; also Field Course: Natural History of the Galapagos Islands (UCLA) Teaching Experience: (Emphases on: Marine Biology, Invertebrate Zoology, Marine Ecology and Natural History, Zoology, Marine Mammals) --Westmont College, 1978-1998, Part time to Full time Instructor --Santa Barbara City College, 1989-1990, Instructor (Human Anatomy) --Westmont College, 1998-2013, Assistant Professor; 2013-Present, Associate Professor --AuSable, Pacific Rim: Assistant/Associate Professor, 1998-2006; 2018-Present --CCSP-New Zealand: Associate Professor, Spring, 2010, Fall, 2011, Fall, 2012 Courses Created/Developed: (Undergraduate level, most Upper Division): Marine Biology, Invertebrate Zoology, Marine Invertebrates, Marine Mammals, Marine Mammal Ecophysiology, Animal Diversity, General Zoology Rank: First Year at Westmont: Instructor (part-Time), 1978; Full-Time, 1988 Previous Rank: Instructor of Biology, 1990-1998; Assistant Professor, 1998-2013 Present Rank: Associate Professor, Fall 2013 to the present Administrative Duties Have Included: