On the Effective Lattice Parameter of Binary Alloys
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Homework 5 Solutions
SCI 1410: materials science & solid state chemistry HOMEWORK 5. solutions Textbook Problems: Imperfections in Solids 1. Askeland 4-67. Why is most “gold” or “silver” jewelry made out of gold or silver alloyed with copper, i.e, what advantages does copper offer? We have two major considerations in jewelry alloying: strength and cost. Copper additions obviously lower the cost of gold or silver (Note that 14K gold is actually only about 50% gold). Copper and other alloy additions also strengthen pure gold and pure silver. In both gold and silver, copper will strengthen the material by either solid solution strengthening (substitutional Cu atoms in the Au or Ag crystal), or by formation of second phase in the microstructure. 2. Solid state solubility. Of the elements in the chart below, name those that would form each of the following relationships with copper (non-metals only have atomic radii listed): a. Substitutional solid solution with complete solubility o Ni and possibly Ag, Pd, and Pt b. Substitutional solid solution of incomplete solubility o Ag, Pd, and Pt (if you didn’t include these in part (a); Al, Co, Cr, Fe, Zn c. An interstitial solid solution o C, H, O (small atomic radii) Element Atomic Radius (nm) Crystal Structure Electronegativity Valence Cu 0.1278 FCC 1.9 +2 C 0.071 H 0.046 O 0.060 Ag 0.1445 FCC 1.9 +1 Al 0.1431 FCC 1.5 +3 Co 0.1253 HCP 1.8 +2 Cr 0.1249 BCC 1.6 +3 Fe 0.1241 BCC 1.8 +2 Ni 0.1246 FCC 1.8 +2 Pd 0.1376 FCC 2.2 +2 Pt 0.1387 FCC 2.2 +2 Zn 0.1332 HCP 1.6 +2 3. -

Chapter 15: Solutions
452-487_Ch15-866418 5/10/06 10:51 AM Page 452 CHAPTER 15 Solutions Chemistry 6.b, 6.c, 6.d, 6.e, 7.b I&E 1.a, 1.b, 1.c, 1.d, 1.j, 1.m What You’ll Learn ▲ You will describe and cate- gorize solutions. ▲ You will calculate concen- trations of solutions. ▲ You will analyze the colliga- tive properties of solutions. ▲ You will compare and con- trast heterogeneous mixtures. Why It’s Important The air you breathe, the fluids in your body, and some of the foods you ingest are solu- tions. Because solutions are so common, learning about their behavior is fundamental to understanding chemistry. Visit the Chemistry Web site at chemistrymc.com to find links about solutions. Though it isn’t apparent, there are at least three different solu- tions in this photo; the air, the lake in the foreground, and the steel used in the construction of the buildings are all solutions. 452 Chapter 15 452-487_Ch15-866418 5/10/06 10:52 AM Page 453 DISCOVERY LAB Solution Formation Chemistry 6.b, 7.b I&E 1.d he intermolecular forces among dissolving particles and the Tattractive forces between solute and solvent particles result in an overall energy change. Can this change be observed? Safety Precautions Dispose of solutions by flushing them down a drain with excess water. Procedure 1. Measure 10 g of ammonium chloride (NH4Cl) and place it in a Materials 100-mL beaker. balance 2. Add 30 mL of water to the NH4Cl, stirring with your stirring rod. -

Self-Assembly of a Colloidal Interstitial Solid Solution with Tunable Sublattice Doping: Supplementary Information
Self-assembly of a colloidal interstitial solid solution with tunable sublattice doping: Supplementary information L. Filion,∗ M. Hermes, R. Ni, E. C. M. Vermolen,† A. Kuijk, C. G. Christova,‡ J. C. P. Stiefelhagen, T. Vissers, A. van Blaaderen, and M. Dijkstra Soft Condensed Matter, Debye Institute for NanoMaterials Science, Utrecht University, Princetonplein 1, NL-3584 CC Utrecht, the Netherlands I. CRYSTAL STRUCTURE LS6 In the main paper we use full free-energy calculations to determine the phase diagram for binary mixtures of hard spheres with size ratio σS/σL = 0.3 where σS(L) is the diameter of the small (large) particles. The phases we considered included a structure with LS6 stoichiometry for which we could not identify an atomic analogue. Snapshots of this structure from various perspectives are shown in Fig. S1. FIG. S1: Unit cell of the binary LS6 superlattice structure described in this paper. Note that both species exhibit long-range crystalline order. The unit cell is based on a body-centered-cubic cell of the large particles in contrast to the face-centered-cubic cell associated with the interstitial solid solution covering most of the phase diagram (Fig. 1). II. PHASE DIAGRAM AT CONSTANT VOLUME 3 In the main paper, we present the xS − p representation of the phase diagram where p = βPσL is the reduced pressure, xS = NS/(NS + NL), NS(L) is the number of small (large) hard spheres, β = 1/kBT , kB is the Boltzmann constant, and T is the absolute temperature. This is the natural representation arising from common tangent construc- tions at constant pressure and the representation required for further simulation studies such as nucleation studies. -

Solid Solution Softening and Enhanced Ductility in Concentrated FCC Silver Solid Solution Alloys
UC Irvine UC Irvine Previously Published Works Title Solid solution softening and enhanced ductility in concentrated FCC silver solid solution alloys Permalink https://escholarship.org/uc/item/7d05p55k Authors Huo, Yongjun Wu, Jiaqi Lee, Chin C Publication Date 2018-06-27 DOI 10.1016/j.msea.2018.05.057 Peer reviewed eScholarship.org Powered by the California Digital Library University of California Materials Science & Engineering A 729 (2018) 208–218 Contents lists available at ScienceDirect Materials Science & Engineering A journal homepage: www.elsevier.com/locate/msea Solid solution softening and enhanced ductility in concentrated FCC silver T solid solution alloys ⁎ Yongjun Huoa,b, , Jiaqi Wua,b, Chin C. Leea,b a Electrical Engineering and Computer Science University of California, Irvine, CA 92697-2660, United States b Materials and Manufacturing Technology University of California, Irvine, CA 92697-2660, United States ARTICLE INFO ABSTRACT Keywords: The major adoptions of silver-based bonding wires and silver-sintering methods in the electronic packaging Concentrated solid solutions industry have incited the fundamental material properties research on the silver-based alloys. Recently, an Solid solution softening abnormal phenomenon, namely, solid solution softening, was observed in stress vs. strain characterization of Ag- Twinning-induced plasticity In solid solution. In this paper, the mechanical properties of additional concentrated silver solid solution phases Localized homologous temperature with other solute elements, Al, Ga and Sn, have been experimentally determined, with their work hardening Advanced joining materials behaviors and the corresponding fractography further analyzed. Particularly, the concentrated Ag-Ga solid so- lution has been discovered to possess the best combination of mechanical properties, namely, lowest yield strength, highest ductility and highest strength, among the concentrated solid solutions of the current study. -

Chapter 18 Solutions and Their Behavior
Chapter 18 Solutions and Their Behavior 18.1 Properties of Solutions Lesson Objectives The student will: • define a solution. • describe the composition of solutions. • define the terms solute and solvent. • identify the solute and solvent in a solution. • describe the different types of solutions and give examples of each type. • define colloids and suspensions. • explain the differences among solutions, colloids, and suspensions. • list some common examples of colloids. Vocabulary • colloid • solute • solution • solvent • suspension • Tyndall effect Introduction In this chapter, we begin our study of solution chemistry. We all might think that we know what a solution is, listing a drink like tea or soda as an example of a solution. What you might not have realized, however, is that the air or alloys such as brass are all classified as solutions. Why are these classified as solutions? Why wouldn’t milk be classified as a true solution? To answer these questions, we have to learn some specific properties of solutions. Let’s begin with the definition of a solution and look at some of the different types of solutions. www.ck12.org 394 E-Book Page 402 Homogeneous Mixtures A solution is a homogeneous mixture of substances (the prefix “homo-” means “same”), meaning that the properties are the same throughout the solution. Take, for example, the vinegar that is used in cooking. Vinegar is approximately 5% acetic acid in water. This means that every teaspoon of vinegar contains 5% acetic acid and 95% water. When a solution is said to have uniform properties, the definition is referring to properties at the particle level. -

Chapter 4 Solution Theory
SMA5101 Thermodynamics of Materials Ceder 2001 Chapter 4 Solution Theory In the first chapters we dealt primarily with closed systems for which only heat and work is transferred between the system and the environment. In the this chapter, we study the thermodynamics of systems that can also exchange matter with other systems or with the environment, and in particular, systems with more than one component. First we focus on homogeneous systems called solutions. Next we consider heterogeneous systems with emphasis on the equilibrium between different multi-component phases. 4.1 WHAT IS A SOLUTION? A solution in thermodynamics refers to a system with more than one chemical component that is mixed homogeneously at the molecular level. A well-known example of a solution is salt water: The Na+, Cl- and H2O ions are intimately mixed at the atomic level. Many systems can be characterized as a dispersion of one phase within another phase. Although such systems typically contain more than one chemical component, they do not form a solution. Solutions are not limited to liquids: for example air, a mixture of predominantly N2 and O2, forms a vapor solution. Solid solutions such as the solid phase in the Si-Ge system are also common. Figure 4.1. schematically illustrates a binary solid solution and a binary liquid solution at the atomic level. Figure 4.1: (a) The (111) plane of the fcc lattice showing a cut of a binary A-B solid solution whereby A atoms (empty circles) are uniformly mixed with B atoms (filled circles) on the atomic level. -

Solid-Liquid Partition Coefficients, Rcjs: What's the Value and When Does It Matter?
Radioprotection - Colloques, volume 37, Cl (2002) Cl-225 Solid-liquid partition coefficients, rCjs: What's the value and when does it matter? M.I. Sheppard and S.C. Sheppard ECOMatters, 24 Aberdeen Avenue, P.O. Box 430, Pinawa, Manitoba ROE 1L0, Canada Abstract. Environmental risk assessments binge on our ability to predict the fete and mobility of radionuclides and metals in terrestrial soils and aquatic sediments. Solid-solution partitioning (the Kd approach), despite its shortcomings, has been used extensively. Much attention has been devoted to grooming the existing key compendia for values applicable for each nuclear risk assessment carried out worldwide. This appears to be an important task. For example, soil Kd values for a single nuclide can vary over several orders of magnitude, yet the soil Kd value is the most important parameter in the soil leaching model. Similarly, plant uptake depends primarily on the nuclide present in solution phase. Despite this apparent sensitivity, our experience has shown that risk assessors dwell too much on the precision of the Kd value for all nuclides. This paper discusses the effect of the Kd value on the resulting soil concentration during leaching and identifies those radionuclides and assessment conditions where a precise value is required. Only those radionuclides that typically have a soil Kd value of 10 LAg or less (Tc, CL I, As) need be accurately described for timeframes of up to 10,000 years for desired soil model prediction outcomes (soil concentrations within two-fold). 1. INTRODUCTION Environmental risk assessments of radionuclides and metals are key to acceptance and sustainability of industrial progress and the disposal of industry's wastes, particularly in the nuclear and imriing and metal smelting industry. -

Chemical Equilibrium and Reaction Modeling of Arsenic and Selenium in Soils
3 Chemical Equilibrium and Reaction Modeling of Arsenic and Selenium in Soils Sabine Goldberg CONTENTS Inorganic Chemistry of As and Se in Soil Solution .......................................... 66 Methylation and Volatilization Reactions ......................................................... 66 Precipitation-Dissolution Reactions .................................................................. 68 Oxidation-Reduction Reactions .......................................................................... 68 Adsorption-Desorption Reactions ..................................................................... 69 Modeling of Adsorption by Soils: Empirical Models ...................................... 70 Modeling of Adsorption by Soils: Constant Capacitance ModeL................ 72 Summary................................................................................................................ 86 References ............................................................................................................... 87 High concentrations of the trace elements arsenic (As) and selenium (Se) in soils pose a threat to agricultural production and the health of humans and animals. As is toxic to both plants and animals. Se, despite being an essential micronutrient for animal nutrition, is potentially toxic because the concentra tion range between deficiency and toxicity in animals is narrow. Seleniferous soils release enough Se to produce vegetation toxic to grazing animals. Such soils occur in the semiarid states of the western United -
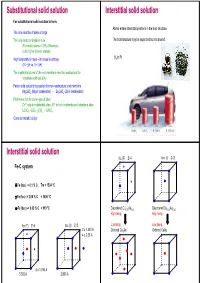
Substitutional Solid Solution Interstitial Solid Solution for Substitutional Solid Solution to Form: Atoms Enters Intersitital Positions in the Host Structure
Substitutional solid solution Interstitial solid solution For substitutional solid solution to form: Atoms enters intersitital positions in the host structure. The ions must be of same charge The ions must be similar in size. The host structure may be expanded but not altered. (For metal atoms < 15% difference) (a bit higher for non-metals) High temperature helps – increase in entropy H2 in Pt (0 > ∆H vs. 0 < ∆H) The crystal structures of the end members must be isostructural for complete solid solubility Partial solid solubility is possible for non-isostructural end members Mg2SiO4 (Mg in octahedras) - Zn2SiO4 (Zn in tetrahedras) Preference for the same type of sites Cr3+ only in octahedral sites, Al3+ in both octahedra and tetraheda sites LiCrO2 -LiCr1-xAlxO2 - LiAlO2 Consider metallic alloys Mg2NH4 LaNi5H6 H2 (liquid) H2 (200 bar) Interstitial solid solution fcc (F) Z=4 bcc (I)Z=2 Fe-C system δ-Fe (bcc) -> 0.1 % C Tm = 1534 °C γ-Fe (fcc) -> 2.06 % C < 1400 °C α-Fe (bcc) -> 0.02 % C < 910 °C Disordered Cu0.75Au0.25 Disordered Cu0.50Au0.50 High temp High temp fcc (F) Z=4 bcc (I)Z=2 Low temp Low temp 2 x 1.433 Å Ordered Cu3Au Ordered CuAu 4 x 2.03 Å 6 x 1.796 Å 3.592 Å 2.866 Å Aliovalent substitution Aliovalent substitution Substitution with ions of different charge 1 Cation vacancies, Substitution by higher valence Need charge compensation mechanism Preserve charge neutrality by leaving out more cations than those that are replaced. Substitution by higher valence cations NaCl dissolves CaCl2 by: Na1-2xCaxVxCl 1 2 Ca2+ vil have a net excess charge of +1 in the structure and attract Na+ vacancies which have net charge -1 Cation vacancies Interstitial anions 2+ 3+ Mg may be replaced by Al : Mg1-3xAl2+2xVxO4 Substitution by lower valence cations 3 4 Anion vacancies Interstitial cations Aliovalent substitution Aliovalent substitution 2 Interstitial anions, Substitution by higher valence 3 Anion vacancies, Substitution by lower valence Preserve charge neutrality by inserting more anions interstitially. -

Modelling the Dissolution/Precipitation of Ideal Solid Solutions
Oil & Gas Science and Technology – Rev. IFP, Vol. 60 (2005), No. 2, pp. 401–415 Copyright © 2005, Institut français du pétrole IFP International Workshop Rencontres scientifiques de l’IFP Gas-Water-Rock Interactions ... / Interactions gaz-eau-roche ... Modelling the Dissolution/Precipitation of Ideal Solid Solutions 1 2 3 4 2 3 E. Nourtier-Mazauric ∗, B. Guy , B. Fritz , E. Brosse , D. Garcia and A. Clément 1 École nationale supérieure des mines de Saint-Etienne – 152 cours Fauriel, 42023 Saint-Etienne Cedex – France 2 idem – affiliated to U.R.A. C.N.R.S. no.6524 3 Centre de géochimie de la surface, C.N.R.S. – 1 rue Blessig, 67084 Strasbourg Cedex – France 4 Institut français du pétrole – 1 et 4 avenue de Bois-Préau, 92852 Rueil-Malmaison Cedex – France e-mail: [email protected] - [email protected] - [email protected] - [email protected] - [email protected] - [email protected] ∗ Corresponding author Résumé — Modélisation de la dissolution/précipitation des solutions solides idéales — Le com- portement cinétique d'une solution solide idéale est modélisé en mettant en compétition deux réactions : la dissolution stoechiométrique du solide existant et la précipitation du composé le moins soluble, c'est- à-dire celui par rapport auquel la sursaturation du fluide est maximum ; les vitesses de ces deux réactions sont exprimées en fonction des écarts à l'équilibre correspondants, les constantes cinétiques dépendant du pH. Dans ce modèle idéal, les courbes de saturation et l'évolution du fluide au sein d'un système réactif simple peuvent être représentées commodément dans des diagrammes en potentiels chimiques. -

A Solution Is a Type of Mixture
¡ ¢ £ ¤ ¥ ¦ § KEY CONCEPT A solution is a type of mixture. BEFORE, you learned NOW, you will learn • Ionic or covalent bonds hold a • How a solution differs from compound together other types of mixtures • Chemical reactions produce • About the parts of a solution chemical changes • How properties of solutions • Chemical reactions alter the differ from properties of their arrangements of atoms separate components VOCABULARY EXPLORE Mixtures solution p. 111 Which substances dissolve in water? solute p. 112 solvent p. 112 PROCEDURE MATERIALS suspension p. 113 • tap water 1 Pour equal amounts of water into each cup. • 2 clear plastic cups 2 Pour one spoonful of table salt into one • plastic spoon of the cups. Stir. • table salt • flour 3 Pour one spoonful of flour into the other cup. Stir. 4 Record your observations. WHAT DO YOU THINK? • Did the salt dissolve? Did the flour dissolve? • How can you tell? The parts of a solution are mixed evenly. VOCABULARY A mixture is a combination of substances, such as a fruit salad. Remember to use the The ingredients of any mixture can be physically separated from each strategy of your choice. You might use a four square other because they are not chemically changed—they are still the same diagram for solution . substances. Sometimes, however, a mixture is so completely blended that its ingredients cannot be identified as different substances. A solution is a type of mixture, called a homogeneous mixture, that is the same throughout. A solution can be physically separated, but all portions of a solution have the same properties. If you stir sand into a glass of water, you can identify the sand as a separate substance that falls to the bottom of the glass. -

Phase Diagrams
Phase diagrams 0.44 wt% of carbon in Fe microstructure of a lead–tin alloy of eutectic composition Primer Materials For Science Teaching Spring 2018 28.6.2018 What is a phase? A phase may be defined as a homogeneous portion of a system that has uniform physical and chemical characteristics Phase Equilibria • Equilibrium is a thermodynamic terms describes a situation in which the characteristics of the system do not change with time but persist indefinitely; that is, the system is stable • A system is at equilibrium if its free energy is at a minimum under some specified combination of temperature, pressure, and composition. The Gibbs Phase Rule This rule represents a criterion for the number of phases that will coexist within a system at equilibrium. Pmax = N + C C – # of components (material that is single phase; has specific stoichiometry; and has a defined melting/evaporation point) N – # of variable thermodynamic parameter (Temp, Pressure, Electric & Magnetic Field) Pmax – maximum # of phase(s) Gibbs Phase Rule – example Phase Diagram of Water C = 1 N = 1 (fixed pressure or fixed temperature) P = 2 fixed pressure (1atm) solid liquid vapor 0 0C 100 0C melting 1 atm. fixed pressure (0.0060373atm=611.73 Pa) boiling Ice Ih vapor 0.01 0C sublimation fixed temperature (0.01 oC) vapor liquid solid 611.73 Pa 109 Pa The Gibbs Phase Rule (2) This rule represents a criterion for the number of degree of freedom within a system at equilibrium. F = N + C - P F – # of degree of freedom: Temp, Pressure, composition (is the number of variables that can be changed independently without altering the phases that coexist at equilibrium) Example – Single Composition 1.