Spectral Terms for Atoms and Molecules
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Coordination Chemistry III: Electronic Spectra
160 Chapter 11 Coordination Chemistry III: Electronic Spectra CHAPTER 11: COORDINATION CHEMISTRY III: ELECTRONIC SPECTRA 11.1 a. p3 There are (6!)/(3!3!) = 20 microstates: MS –3/2 –1/2 1/2 3/2 + – – + – + +2 1 1 0 1 1 0 + – – + – + 1 1 –1 1 1 –1 +1 – – + + – + 1 0 0 1 0 0 + – – + + – 1 0 –1 1 0 –1 – – – – + – + – + + + + ML 0 1 0 –1 1 0 –1 1 0 –1 1 0 –1 1– 0– –1+ 1– 0+ –1+ + – – + – + –1 –1 1 –1 –1 1 –1 – – + + – + –1 0 0 –1 0 0 + – – + – + –2 –1 –1 0 –1 –1 0 Terms: L = 0, S = 3/2: 4S (ground state) L = 2, S = 1/2: 2D 2 L = 1, S = 1/2: P 6! 10! b. p1d1 There are 60 microstates: 1!5! 1!9! M S –1 0 1 – – – + + – + + 3 1 2 1 2 , 1 2 1 2 – – + – – + + + 1 1 1 1 , 1 1 1 1 2 + + 0– 2– 0– 2+, 0+ 2– 0 2 1– 0– 1+ 0–, 0+ 1– 1+ 0+ + + 1 0– 1– 1– 0+, 0– 1+ 0 1 + + –1– 2– –1– 2+, –1+ 2– –1 2 – – + – – + + + 1 –1 1 –1 , 1 –1 1 –1 – – – + + – + + ML 0 0 0 0 0 , 0 0 0 0 + + –1– 1– –1+ 1–, –1– 1+ –1 1 – – + – + – + + –1 0 –1 0 , 0 –1 –1 0 – – – + – + + + –1 0 –1 –1 0 , 0 –1 0 –1 – – – + + – 1+ –2+ 1 –2 1 –2 , 1 –2 –1– –1– –1+ –1–, –1– –1+ –1– –1+ –2 + – – + + + 0– –2– 0 –2 , 0 –2 0 –2 –3 –1– –2– –1– –2+, –1+ –2– –1+ –2+ Terms: L = 3, S = 1 3F (ground state) L = 2, S = 0 1D L = 3, S = 0 1F L = 1, S = 1 3P 3 1 L = 2, S = 1 D L = 1, S = 0 P The two electrons have quantum numbers that are independent of each other, because the electrons are in different orbitals. -

Review Sheet on Determining Term Symbols
Review Sheet on Determining Term Symbols Term symbols for electronic configurations are useful not only to the spectroscopist but also to the inorganic chemist interested in understanding electronic and magnetic properties of molecules. We will concentrate on the method of Douglas and McDaniel (p. 26ff); however, you may find other treatments equal or superior to the D & M method. Term symbols are a shorthand method used to describe the energy, angular momentum, and spin multiplicity of an atom in any particular state. The general form is a given as Tj where T is a capital letter corresponding to the value of L (the angular momentum quantum number) and may be assigned as S, P, D, F, G, … for |L| = 0, 1, 2, 3, 4, … respectively. The superscript “a” is called the spin multiplicity and can be evaluated as a = 2S +1 where S is the spin quantum number. The subscript “j” is the numerical value of J, a new quantum number defined as: J = l +S, which corresponds to the total orbital and spin angular momentum of the system. The term symbol 3P is read as triplet – Pee state and indicates that there are two unpaired electrons in a state with maximum orbital angular momentum, L=1. The number of microstates (N) of a system corresponds to the total number of distinct arrangements for “e” number of electrons to be placed in “n” number of possible orbital positions. N = # of microstates = n!/(e!(n-e)!) For a set of p orbitals n = 6 since there are 2 positions in each orbital. -

XSAMS: XML Schema for Atomic, Molecular and Solid Data
XSAMS: XML Schema for Atomic, Molecular and Solid Data Version 0.1.1 Draft Document, January 14, 2011 This version: http://www-amdis.iaea.org/xsams/docu/v0.1.1.pdf Latest version: http://www-amdis.iaea.org/xsams/docu/ Previous versions: http://www-amdis.iaea.org/xsams/docu/v0.1.pdf Editors: M.L. Dubernet, D. Humbert, Yu. Ralchenko Authors: M.L. Dubernet, D. Humbert, Yu. Ralchenko, E. Roueff, D.R. Schultz, H.-K. Chung, B.J. Braams Contributors: N. Moreau, P. Loboda, S. Gagarin, R.E.H. Clark, M. Doronin, T. Marquart Abstract This document presents a proposal for an XML schema aimed at describing Atomic, Molec- ular and Particle Surface Interaction Data in distributed databases around the world. This general XML schema is a collaborative project between International Atomic Energy Agency (Austria), National Institute of Standards and Technology (USA), Universit´ePierre et Marie Curie (France), Oak Ridge National Laboratory (USA) and Paris Observatory (France). Status of this document It is a draft document and it may be updated, replaced, or obsoleted by other documents at any time. It is inappropriate to use this document as reference materials or to cite them as other than “work in progress”. Acknowledgements The authors wish to acknowledge all colleagues and institutes, atomic and molecular database experts and physicists who have collaborated through different discussions to the building up of the concepts described in this document. Change Log Version 0.1: June 2009 Version 0.1.1: November 2010 Contents 1 Introduction 11 1.1 Motivation..................................... 11 1.2 Limitations..................................... 12 2 XSAMS structure, types and attributes 13 2.1 Atomic, Molecular and Particle Surface Interaction Data XML Schema Structure 13 2.2 Attributes .................................... -

Atomic Spectroscopy Summary
ATOMIC SPECTROSCOPY SUMMARY TERM SYMBOLS Because the total angular momentum and energy commute, we can use the angular momentum (L) to label the energy states. Note that electron configurations are ambiguous in that we don’t specify which orbital or which spin each electron has. This means that there are a number of different sets of 푚ℓ and 푚푠 that are consistent with a given configuration. Why are there different energies? Because of electron/electron repulsion and spin-orbit coupling, the ways we can fill the orbitals DO NOT NESCESSARILLY have the same energy. (Remember that we talked about how Hund’s rules come from this. How spin up elelctrons interact less with other spin up electrons in the same subshell.) Components of a Term Symbol: 2푆+1 퐿퐽 RULES FOR TERM SYMBOLS Term Symbols are assigned based on two main rules: 1. Unsöld’s Rule: All filled shells/subshells are ignored when calculating L and S. Basically, all of the electrons will have their contributions to spin and orbital angular momentum canceled by other electrons in the same subshell. 2. “holes”: The state of the “hole” (an empty spot in an orbital where an electron could be but isn’t) is the same as the state of an electron. This means that when we look at a p5 we can pretend it is a p1. This is incredibly useful as it means that instead of 5 electrons to tinker with, you can just act like it is 1. CALCULATING L: L is the Total Angular Momentum L= (sum of ℓ values) ….(smallest positive difference of ℓ values) o 2 s electrons (different shells) L=0+0,…,0-0 L=0 o 1 d electron and 1 p electron L=2+1,…,2-1 L=3, 2, 1 S is the Total Spin S is the total spin quantum number of the electrons in an atom. -

Chapter 8 2013
described by Core Chapter 8 Electrons Wave Function e- filling spdf electronic configuration comprising (Orbital) Valence determined by Electrons described by Aufbau basis for Rules Periodic Quantum which involve Table Numbers which summarizes Orbital Pauli Hund’s which are Energy Exclusion Rule Periodic Properties Electron Configuration and Chemical Periodicity Quantum 4-quantum numbers specify the energy and location of Numbers electrons around a nucleus (all we can know). This numbers are the framework for the “electronic structure Orbital size of an atom”. Principal define & energy n = 1,2,3,.. s defines Name Symbol Permitted Values Property principal n positive integers (1,2,3...) orbital energy (size) Angular Orbital angular l orbital shape (0, 1, 2, and 3 momentum, l defines momentum integers from 0 to n-1 correspond to s, p, d, and f shape orbitals, respectively.) Magnetic Orbital magnetic integers from -l to 0 to +l orbital orientation in space defines ml ml orientation direction of e- spin spin m +1/2 or -1/2 Electron s Spin, ms defines spin Quantum Allowed Schrodinger’s equation gives an exact solution for H- Possible Orbitals Number Values atom, but does not for many electron-atoms. Electron- n Positive integers 1 2 3 electron repulsion in multi-electron split energy levels. 1,2,3,4.... Hydrogen Atom Multi-electron atoms 0 0 1 0 1 2 l 0 up to max Orbitals are of n-1 “degenerate” or Energy the same energy in Hydrogen! ml -l,...0...+l 0 0 -1 0 1 0 -1 0 1 -2-1 0 12 Orbital Name 1s 2s 2p 3s 3p 3d Electrons will Shapes or Boundry fill lowest Surface Plots energy orbitals first! Inner core electrons “shield” or “screen” outer 4-quantum numbers specify all the information we can know the energy electrons from the positive charge of the nucleus. -

Review of Term Symbols Computation from Multi-Electron Systems for Chemistry Students Jamiu A
S O s Contemporary Chemistry p e s n Acce REVIEW ARTICLE Review of Term Symbols Computation from Multi-Electron Systems for Chemistry Students Jamiu A. Odutola* Department of Physics, Chemistry and Mathematics, Alabama Agricultural and Mechanical University, Normal AL 35762, USA Abstract We have computed the term symbols resulting from the coupling of angular momenta of both spin and orbital from the ground state electron configuration. Configurations of equivalent electrons present challenges and were handled by the group theoretical methods. Terms are the combined values of L and S while the combined values of L, S and J defines the level. Key Words: Terms, Term Symbols, Spectroscopic term symbols, State, Level, Sub-level, Equivalent electrons, Non-equivalent electrons, Orbital angular momentum, Spin angular momentum, Spin-Orbital angular momentum, Russell-Saunders Coupling, LS coupling, j-j coupling and multiplicity. Introduction four quantum numbers (n, l, ml, ms). The first three quantum numbers in the set describes the spatial distribution and the Spectroscopic term symbols are very important in classifying last describes the spin state of the electron. n is the principal the electronic states of atoms and molecules particularly in quantum number and it describes the size or the energy level physical and inorganic chemistry [1-4]. There is a vast amount of the shell (or atom). l is the azimuthal or angular momentum of information on the topic of term symbols and techniques quantum number and it describes the shape of the orbital for obtaining these terms in the literature. Many authors gave or the subshell. m is the magnetic quantum number and it credit to such textbooks as Cotton and Wilkinson [5] as well as l describes the spatial orientation or direction of the orbital. -

Ultrasoft Pseudopotentials for Lanthanide Solvation Complexes: Core Or Valence Character of the 4F Electrons Rodolphe Pollet, C
Ultrasoft pseudopotentials for lanthanide solvation complexes: Core or valence character of the 4f electrons Rodolphe Pollet, C. Clavaguéra, J.-P. Dognon To cite this version: Rodolphe Pollet, C. Clavaguéra, J.-P. Dognon. Ultrasoft pseudopotentials for lanthanide solvation complexes: Core or valence character of the 4f electrons. Journal of Chemical Physics, American Institute of Physics, 2006, 124, pp.164103 - 1-6. 10.1063/1.2191498. hal-00083928 HAL Id: hal-00083928 https://hal.archives-ouvertes.fr/hal-00083928 Submitted on 21 Jul 2006 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Ultrasoft pseudopotentials for lanthanide solvation complexes: Core or valence character of the 4f electrons Rodolphe Pollet,∗ Carine Clavagu´era, Jean-Pierre Dognon Theoretical Chemistry Laboratory DSM/DRECAM/SPAM-LFP (CEA-CNRS URA2453) CEA/SACLAY, Bat. 522, 91191 GIF SUR YVETTE, FRANCE March 6, 2006 Abstract The 4f electrons of lanthanides, because of their strong localiza- tion in the region around the nucleus, are traditionally included in a pseudopotential core. This approximation is scrutinized by optimizing 3+ the structures and calculating the interaction energies of Gd (H2O) 3+ and Gd (NH3) microsolvation complexes within plane wave PBE cal- culations using ultrasoft pseudopotentials where the 4f electrons are included either in the core or in the valence space. -

Chapter 7 Periodic Properties of the Elements Learning Outcomes
Chapter 7 Periodic Properties of the Elements Learning Outcomes: Explain the meaning of effective nuclear charge, Zeff, and how Zeff depends on nuclear charge and electron configuration. Predict the trends in atomic radii, ionic radii, ionization energy, and electron affinity by using the periodic table. Explain how the radius of an atom changes upon losing electrons to form a cation or gaining electrons to form an anion. Write the electron configurations of ions. Explain how the ionization energy changes as we remove successive electrons, and the jump in ionization energy that occurs when the ionization corresponds to removing a core electron. Explain how irregularities in the periodic trends for electron affinity can be related to electron configuration. Explain the differences in chemical and physical properties of metals and nonmetals, including the basicity of metal oxides and the acidity of nonmetal oxides. Correlate atomic properties, such as ionization energy, with electron configuration, and explain how these relate to the chemical reactivity and physical properties of the alkali and alkaline earth metals (groups 1A and 2A). Write balanced equations for the reactions of the group 1A and 2A metals with water, oxygen, hydrogen, and the halogens. List and explain the unique characteristics of hydrogen. Correlate the atomic properties (such as ionization energy, electron configuration, and electron affinity) of group 6A, 7A, and 8A elements with their chemical reactivity and physical properties. Development of Periodic Table •Dmitri Mendeleev and Lothar Meyer (~1869) independently came to the same conclusion about how elements should be grouped in the periodic table. •Henry Moseley (1913) developed the concept of atomic numbers (the number of protons in the nucleus of an atom) 1 Predictions and the Periodic Table Mendeleev, for instance, predicted the discovery of germanium (which he called eka-silicon) as an element with an atomic weight between that of zinc and arsenic, but with chemical properties similar to those of silicon. -

Synthesis, Structure and Bonding of Actinide Disulphide Dications in the Gas Phase
Synthesis, structure and bonding of actinide disulphide dications in the gas phase Ana F. Lucena,1 Nuno A. G. Bandeira,2,3,4,* Cláudia C. L. Pereira,1,§ John K. Gibson,5 Joaquim Marçalo1,* 1 Centro de Ciências e Tecnologias Nucleares, Instituto Superior Técnico, Universidade de Lisboa, 2695- 066 Bobadela LRS, Portugal 2 Institute of Chemical Research of Catalonia (ICIQ), Barcelona Institute of Technology (BIST), 16 – Av. Països Catalans, 43007 Tarragona, Spain 3 Centro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais 1, 1049-001 Lisboa, Portugal 4 Centro de Química e Bioquímica, Faculdade de Ciências, Universidade de Lisboa, Campo Grande, 1749-016 Lisboa, Portugal 5 Chemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA § Present address: REQUIMTE, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, 2829-516 Caparica, Portugal * Corresponding authors: Nuno A. G. Bandeira, email: [email protected]; Joaquim Marçalo, email: [email protected] Abstract 2+ Actinide disulphide dications, AnS2 , were produced in the gas phase for An = Th and Np by reaction of An2+ cations with the sulfur-atom donor COS, in a sequential abstraction process of two sulfur atoms, as examined by FTICR mass spectrometry. For An = Pu and Am, An2+ ions were unreactive with COS and did not yield any sulphide species. High level multiconfigurational (CASPT2) calculations were performed to assess the structures and 2+ bonding of the new AnS2 species obtained for An = Th, Np, as well as for An = Pu to examine trends along the An series, and for An = U to compare with a previous experimental study and 2+ DFT computational scrutiny of US2 . -
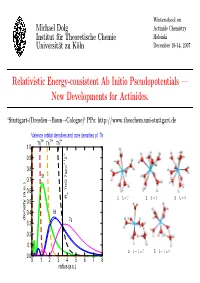
Relativistic Energy-Consistent Ab Initio Pseudopotentials — New Developments for Actinides
Winterschool on Michael Dolg Actinide Chemistry Institut fur¨ Theoretische Chemie Helsinki Universit¨at zu K¨oln December 10-14, 2007 Relativistic Energy-consistent Ab Initio Pseudopotentials — New Developments for Actinides. ‘Stuttgart-(Dresden→Bonn→Cologne)‘ PPs: http://www.theochem.uni-stuttgart.de Valence orbital densities and core densities of Th 30+ 12+ 4+ Th Th Th 1.0 + 0.9 Σ 2 1 0.8 σ 5f 0.7 0.6 ThO 7sp e R 1 h =7 2 h =8 3 h =9 0.5 0.4 6d density (a.u.) 7s 0.3 0.2 0.1 4 h − 1=7 5 h − 1=8 0.0 0 1 2 3 4 5 6 7 8 radius (a.u.) Contents • Short introduction to energy-consistent relativistic pseudopotentials • What we currently can provide for lanthanides and actinides: Wood-Boring adjusted scalar-relativistic (one-component) large-core (Ln 4f-in-core, An 5f-in-core) and small-core (Ln 4f-in-valence, An 5f-in-valence) pseudopotentials, valence spin-orbit operators in the small core case and corresponding valence basis sets of different quality (VDZ to VQZ) and contraction schemes (segmented, generalized) based on ANOs. – some general considerations concerning the choice of the core: small-core vs. large-core PPs; f-in-core PPs (superconfiguration model (R.W.Field); feudal model (B.Bursten) ?) – calibration for atoms/molecules – examples for applications • What we would like to have for lanthanides and actinides: Multi-configuration Dirac-Hartree-Fock/Dirac-Coulomb-Breit adjusted relativistic (two- and one-component) small-core pseudopotentials and corresponding correlation- consistent valence basis sets. -

Alkali Metal Spectra
____________________________________________________________________________________________________ Subject Chemistry Paper No and Title 8 and Physical Spectroscopy Module No and Title 8: Alkali metal spectra Module Tag CHE_P8_M8 CHEMISTRY PAPER No. : 8 (PHYSICAL SPECTROSCOPY) MODULE No. : 8 (ALKALI METAL SPECTRA) ____________________________________________________________________________________________________ TABLE OF CONTENTS 1. Learning Outcomes 2. Introduction 3. Multi-electron Atoms 4. Electron Orbitals 5. Alkali Metal Spectra 6. Summary CHEMISTRY PAPER No. : 8 (PHYSICAL SPECTROSCOPY) MODULE No. : 8 (ALKALI METAL SPECTRA) ____________________________________________________________________________________________________ 1. Learning Outcomes In this module, you will study about the failures of the Bohr theory, which led to the formulation of the quantum theory. You will understand how the new theory could explain the fine structure in the spectra of hydrogen and hydrogen-like ions, and how this theory can be extended to atoms which have a single electron in their outermost shell, i.e. the alkali metal atoms. You should also be able to write the term symbols for simple one-electron systems. 2. Introduction Bohr’s model could only explain the line spectra of hydrogen and hydrogen-like ions. However, it did not even attempt to explain the spectra of multi-electron atoms. Even in the case of hydrogen, higher resolution shows that each line is split into a doublet, which the Bohr theory was clearly unable to explain. Clearly, the Bohr theory was inadequate and a better, universally applicable, theory was required. 3. Multi-electron Atoms The problem with multi-electron atoms is that, not only are there several electrons getting attracted to the same nucleus, they are repelling each other as well. This repulsion is much harder to deal with. -

QMEPC Discovery of Light's Medium Explains Why Gravity Is Larger and Weaker Odin Von Aesir* PO Box 50252, Austin, TX 78763, 925-202-6631, USA
Research & Reviews: Journal of Pure and Applied Physics QMEPC Discovery of Light's Medium Explains Why Gravity Is Larger and Weaker Odin Von Aesir* PO Box 50252, Austin, TX 78763, 925-202-6631, USA Review Article Received date: 17/11/2015 ABSTRACT Accepted date: 25/02/2016 The MEPC – The Magnetic Electron Plasma Cloud – is plasma solely Published date: 30/03/2016 composed of electrons that is the source and recipient of electrons that exist outside of the atom. The ramifications of this are nothing short of *For Correspondence phenomenal, because it not only explains where electrons come from and how they are available for distribution around the atomic nucleus, but it also Odin Von Aesir, Po Box 50252, Austin, TX explains: 78763, 925-202-6631, USA • What occupies the space in between the atoms? E-mail: [email protected] • The existence of a hierarchy of forces surrounding the atomic core, • Why gravity is larger and weaker than the other known forces, Keywords: Ramifications; Hierarchy; Proximity; • What the medium of light is, Stratosphere • Why light is both a particle and a wave, and • What dark matter and dark energy are? INTRODUCTION Furthermore, the MEPC makes logical sense of the quirkiness of quantum mechanics and the progression of time and space, because it explains the composition and connection of everything. It even gives an insight into the makeup of the atomic nucleus itself [1]. It is common knowledge that the space that surrounds us and that separates us from each other and other things here on earth is composed of air molecules that are made up of even smaller atoms.