NGS Data and Sequence Alignment
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

File Formats Exercises
Introduction to high-throughput sequencing file formats – advanced exercises Daniel Vodák (Bioinformatics Core Facility, The Norwegian Radium Hospital) ( [email protected] ) All example files are located in directory “/data/file_formats/data”. You can set your current directory to that path (“cd /data/file_formats/data”) Exercises: a) FASTA format File “Uniprot_Mammalia.fasta” contains a collection of mammalian protein sequences obtained from the UniProt database ( http://www.uniprot.org/uniprot ). 1) Count the number of lines in the file. 2) Count the number of header lines (i.e. the number of sequences) in the file. 3) Count the number of header lines with “Homo” in the organism/species name. 4) Count the number of header lines with “Homo sapiens” in the organism/species name. 5) Display the header lines which have “Homo” in the organism/species names, but only such that do not have “Homo sapiens” there. b) FASTQ format File NKTCL_31T_1M_R1.fq contains a reduced collection of tumor read sequences obtained from The Sequence Read Archive ( http://www.ncbi.nlm.nih.gov/sra ). 1) Count the number of lines in the file. 2) Count the number of lines beginning with the “at” symbol (“@”). 3) How many reads are there in the file? c) SAM format File NKTCL_31T_1M.sam contains alignments of pair-end reads stored in files NKTCL_31T_1M_R1.fq and NKTCL_31T_1M_R2.fq. 1) Which program was used for the alignment? 2) How many header lines are there in the file? 3) How many non-header lines are there in the file? 4) What do the non-header lines represent? 5) Reads from how many template sequences have been used in the alignment process? c) BED format File NKTCL_31T_1M.bed holds information about alignment locations stored in file NKTCL_31T_1M.sam. -

EMBL-EBI Powerpoint Presentation
Processing data from high-throughput sequencing experiments Simon Anders Use-cases for HTS · de-novo sequencing and assembly of small genomes · transcriptome analysis (RNA-Seq, sRNA-Seq, ...) • identifying transcripted regions • expression profiling · Resequencing to find genetic polymorphisms: • SNPs, micro-indels • CNVs · ChIP-Seq, nucleosome positions, etc. · DNA methylation studies (after bisulfite treatment) · environmental sampling (metagenomics) · reading bar codes Use cases for HTS: Bioinformatics challenges Established procedures may not be suitable. New algorithms are required for · assembly · alignment · statistical tests (counting statistics) · visualization · segmentation · ... Where does Bioconductor come in? Several steps: · Processing of the images and determining of the read sequencest • typically done by core facility with software from the manufacturer of the sequencing machine · Aligning the reads to a reference genome (or assembling the reads into a new genome) • Done with community-developed stand-alone tools. · Downstream statistical analyis. • Write your own scripts with the help of Bioconductor infrastructure. Solexa standard workflow SolexaPipeline · "Firecrest": Identifying clusters ⇨ typically 15..20 mio good clusters per lane · "Bustard": Base calling ⇨ sequence for each cluster, with Phred-like scores · "Eland": Aligning to reference Firecrest output Large tab-separated text files with one row per identified cluster, specifying · lane index and tile index · x and y coordinates of cluster on tile · for each -

Sequence Alignment/Map Format Specification
Sequence Alignment/Map Format Specification The SAM/BAM Format Specification Working Group 3 Jun 2021 The master version of this document can be found at https://github.com/samtools/hts-specs. This printing is version 53752fa from that repository, last modified on the date shown above. 1 The SAM Format Specification SAM stands for Sequence Alignment/Map format. It is a TAB-delimited text format consisting of a header section, which is optional, and an alignment section. If present, the header must be prior to the alignments. Header lines start with `@', while alignment lines do not. Each alignment line has 11 mandatory fields for essential alignment information such as mapping position, and variable number of optional fields for flexible or aligner specific information. This specification is for version 1.6 of the SAM and BAM formats. Each SAM and BAMfilemay optionally specify the version being used via the @HD VN tag. For full version history see Appendix B. Unless explicitly specified elsewhere, all fields are encoded using 7-bit US-ASCII 1 in using the POSIX / C locale. Regular expressions listed use the POSIX / IEEE Std 1003.1 extended syntax. 1.1 An example Suppose we have the following alignment with bases in lowercase clipped from the alignment. Read r001/1 and r001/2 constitute a read pair; r003 is a chimeric read; r004 represents a split alignment. Coor 12345678901234 5678901234567890123456789012345 ref AGCATGTTAGATAA**GATAGCTGTGCTAGTAGGCAGTCAGCGCCAT +r001/1 TTAGATAAAGGATA*CTG +r002 aaaAGATAA*GGATA +r003 gcctaAGCTAA +r004 ATAGCT..............TCAGC -r003 ttagctTAGGC -r001/2 CAGCGGCAT The corresponding SAM format is:2 1Charset ANSI X3.4-1968 as defined in RFC1345. -

Introduction to Bioinformatics (Elective) – SBB1609
SCHOOL OF BIO AND CHEMICAL ENGINEERING DEPARTMENT OF BIOTECHNOLOGY Unit 1 – Introduction to Bioinformatics (Elective) – SBB1609 1 I HISTORY OF BIOINFORMATICS Bioinformatics is an interdisciplinary field that develops methods and software tools for understanding biologicaldata. As an interdisciplinary field of science, bioinformatics combines computer science, statistics, mathematics, and engineering to analyze and interpret biological data. Bioinformatics has been used for in silico analyses of biological queries using mathematical and statistical techniques. Bioinformatics derives knowledge from computer analysis of biological data. These can consist of the information stored in the genetic code, but also experimental results from various sources, patient statistics, and scientific literature. Research in bioinformatics includes method development for storage, retrieval, and analysis of the data. Bioinformatics is a rapidly developing branch of biology and is highly interdisciplinary, using techniques and concepts from informatics, statistics, mathematics, chemistry, biochemistry, physics, and linguistics. It has many practical applications in different areas of biology and medicine. Bioinformatics: Research, development, or application of computational tools and approaches for expanding the use of biological, medical, behavioral or health data, including those to acquire, store, organize, archive, analyze, or visualize such data. Computational Biology: The development and application of data-analytical and theoretical methods, mathematical modeling and computational simulation techniques to the study of biological, behavioral, and social systems. "Classical" bioinformatics: "The mathematical, statistical and computing methods that aim to solve biological problems using DNA and amino acid sequences and related information.” The National Center for Biotechnology Information (NCBI 2001) defines bioinformatics as: "Bioinformatics is the field of science in which biology, computer science, and information technology merge into a single discipline. -

NGS Raw Data Analysis
NGS raw data analysis Introduc3on to Next-Generaon Sequencing (NGS)-data Analysis Laura Riva & Maa Pelizzola Outline 1. Descrip7on of sequencing plaorms 2. Alignments, QC, file formats, data processing, and tools of general use 3. Prac7cal lesson Illumina Sequencing Single base extension with incorporaon of fluorescently labeled nucleodes Library preparaon Automated cluster genera3on DNA fragmenta3on and adapter ligaon Aachment to the flow cell Cluster generaon by bridge amplifica3on of DNA fragments Sequencing Illumina Output Quality Scores Sequence Files Comparison of some exisng plaGorms 454 Ti RocheT Illumina HiSeqTM ABI 5500 (SOLiD) 2000 Amplificaon Emulsion PCR Bridge PCR Emulsion PCR Sequencing Pyrosequencing Reversible Ligaon-based reac7on terminators sequencing Paired ends/sep Yes/3kb Yes/200 bp Yes/3 kb Read length 400 bp 100 bp 75 bp Advantages Short run 7mes. The most popular Good base call Longer reads plaorm accuracy. Good improve mapping in mulplexing repe7ve regions. capability Ability to detect large structural variaons Disadvantages High reagent cost. Higher error rates in repeat sequences The Illumina HiSeq! Stacey Gabriel Libraries, lanes, and flowcells Flowcell Lanes Illumina Each reaction produces a unique Each NGS machine processes a library of DNA fragments for single flowcell containing several sequencing. independent lanes during a single sequencing run Applications of Next-Generation Sequencing Basic data analysis concepts: • Raw data analysis= image processing and base calling • Understanding Fastq • Understanding Quality scores • Quality control and read cleaning • Alignment to the reference genome • Understanding SAM/BAM formats • Samtools • Bedtools Understanding FASTQ file format FASTQ format is a text-based format for storing a biological sequence and its corresponding quality scores. -

Large Scale Genomic Rearrangements in Selected
Pucker et al. BMC Genomics (2021) 22:599 https://doi.org/10.1186/s12864-021-07877-8 RESEARCH ARTICLE Open Access Large scale genomic rearrangements in selected Arabidopsis thaliana T-DNA lines are caused by T-DNA insertion mutagenesis Boas Pucker1,2† , Nils Kleinbölting3† and Bernd Weisshaar1* Abstract Background: Experimental proof of gene function assignments in plants is based on mutant analyses. T-DNA insertion lines provided an invaluable resource of mutants and enabled systematic reverse genetics-based investigation of the functions of Arabidopsis thaliana genes during the last decades. Results: We sequenced the genomes of 14 A. thaliana GABI-Kat T-DNA insertion lines, which eluded flanking sequence tag-based attempts to characterize their insertion loci, with Oxford Nanopore Technologies (ONT) long reads. Complex T-DNA insertions were resolved and 11 previously unknown T-DNA loci identified, resulting in about 2 T-DNA insertions per line and suggesting that this number was previously underestimated. T-DNA mutagenesis caused fusions of chromosomes along with compensating translocations to keep the gene set complete throughout meiosis. Also, an inverted duplication of 800 kbp was detected. About 10 % of GABI-Kat lines might be affected by chromosomal rearrangements, some of which do not involve T-DNA. Local assembly of selected reads was shown to be a computationally effective method to resolve the structure of T-DNA insertion loci. We developed an automated workflow to support investigation of long read data from T-DNA insertion lines. All steps from DNA extraction to assembly of T-DNA loci can be completed within days. Conclusions: Long read sequencing was demonstrated to be an effective way to resolve complex T-DNA insertions and chromosome fusions. -

L3: Short Read Alignment to a Reference Genome
L3: Short Read Alignment to a Reference Genome Shamith Samarajiwa CRUK Autumn School in Bioinformatics Cambridge, September 2017 Where to get help! http://seqanswers.com http://www.biostars.org http://www.bioconductor.org/help/mailing-list Read the posting guide before sending email! Overview ● Understand the difference between reference genome builds ● Introduction to Illumina sequencing ● Short read aligners ○ BWA ○ Bowtie ○ STAR ○ Other aligners ● Coverage and Depth ● Mappability ● Use of decoy and sponge databases ● Alignment Quality, SAMStat, Qualimap ● Samtools and Picard tools, ● Visualization of alignment data ● A very brief look at long reads, graph genome aligners and de novo genome assembly Reference Genomes ● A haploid representation of a species genome. ● The human genome is a haploid mosaic derived from 13 volunteer donors from Buffalo, NY. USA. ● In regions where there is known large scale population variation, sets of alternate loci (178 in GRCh38) are assembled alongside the reference locus. ● The current build has around 500 gaps, whereas the first version had ~150,000 gaps. Genome Reference Consortium: https://www.ncbi.nlm.nih.gov/grc £600 30X Sequencers http://omicsmap.com Illumina sequencers Illumina Genome Analyzer Illumina sequencing technology ● Illumina sequencing is based on the Solexa technology developed by Shankar Balasubramanian and David Klenerman (1998) at the University of Cambridge. ● Multiple steps in “Sequencing by synthesis” (explained in next slide) ○ Library Preparation ○ Bridge amplification and Cluster generation ○ Sequencing using reversible terminators ○ Image acquisition and Fastq generation ○ Alignment and data analysis Illumina Flowcell Sequencing By Synthesis technology Illumina Sequencing Bridge Amplification Sequencing Cluster growth Incorporation of fluorescence, reversibly terminated tagged nt Multiplexing • Multiplexing gives the ability to sequence multiple samples at the same time. -

Annual Scientific Report 2011 Annual Scientific Report 2011 Designed and Produced by Pickeringhutchins Ltd
European Bioinformatics Institute EMBL-EBI Annual Scientific Report 2011 Annual Scientific Report 2011 Designed and Produced by PickeringHutchins Ltd www.pickeringhutchins.com EMBL member states: Austria, Croatia, Denmark, Finland, France, Germany, Greece, Iceland, Ireland, Israel, Italy, Luxembourg, the Netherlands, Norway, Portugal, Spain, Sweden, Switzerland, United Kingdom. Associate member state: Australia EMBL-EBI is a part of the European Molecular Biology Laboratory (EMBL) EMBL-EBI EMBL-EBI EMBL-EBI EMBL-European Bioinformatics Institute Wellcome Trust Genome Campus, Hinxton Cambridge CB10 1SD United Kingdom Tel. +44 (0)1223 494 444, Fax +44 (0)1223 494 468 www.ebi.ac.uk EMBL Heidelberg Meyerhofstraße 1 69117 Heidelberg Germany Tel. +49 (0)6221 3870, Fax +49 (0)6221 387 8306 www.embl.org [email protected] EMBL Grenoble 6, rue Jules Horowitz, BP181 38042 Grenoble, Cedex 9 France Tel. +33 (0)476 20 7269, Fax +33 (0)476 20 2199 EMBL Hamburg c/o DESY Notkestraße 85 22603 Hamburg Germany Tel. +49 (0)4089 902 110, Fax +49 (0)4089 902 149 EMBL Monterotondo Adriano Buzzati-Traverso Campus Via Ramarini, 32 00015 Monterotondo (Rome) Italy Tel. +39 (0)6900 91402, Fax +39 (0)6900 91406 © 2012 EMBL-European Bioinformatics Institute All texts written by EBI-EMBL Group and Team Leaders. This publication was produced by the EBI’s Outreach and Training Programme. Contents Introduction Foreword 2 Major Achievements 2011 4 Services Rolf Apweiler and Ewan Birney: Protein and nucleotide data 10 Guy Cochrane: The European Nucleotide Archive 14 Paul Flicek: -

Efficient Synchronization of Paired-End Fastq Files
bioRxiv preprint doi: https://doi.org/10.1101/552885; this version posted February 19, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Fastq-pair: efficient synchronization of paired-end fastq files. 1 2,* John A. Edwards and Robert A. Edwards 1 Sigma Numerix Ltd. Coalville, Leicestershire, England 2 San Diego State University, 5500 Campanile Drive, San Diego, CA 92182 * Corresponding author Dr. Robert Edwards Department of Computer Science San Diego State University 5500 Campanile Dr San Diego, CA 92182 Email: [email protected] Tel: 619 594 1672 Keywords: fastq, next generation sequencing bioRxiv preprint doi: https://doi.org/10.1101/552885; this version posted February 19, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Abstract Paired end DNA sequencing provides additional information about the sequence data that is used in sequence assembly, mapping, and other downstream bioinformatics analysis. Paired end reads are usually provided as two fastq-format files, with each file representing one end of the read. Many commonly used downstream tools require that the sequence reads appear in each file in the same order, and reads that do not have a pair in the corresponding file are placed in a separate file of singletons. -
Database Generation
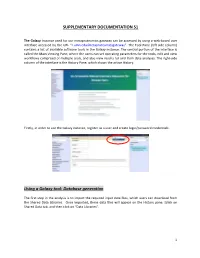
SUPPLEMENTARY DOCUMENTATION S1 The Galaxy Instance used for our metaproteomics gateway can be accessed by using a web-based user interface accessed by the URL “z.umn.edu/metaproteomicsgateway”. The Tool Pane (left side column) contains a list of available software tools in the Galaxy instance. The central portion of the interface is called the Main Viewing Pane, where the users can set operating parameters for the tools, edit and view worKflows comprised of multiple tools, and also view results for and from data analyses. The right-side column of the interface is the History Pane, which shows the active History. Firstly, in order to use the Galaxy instance, register as a user and create login/password credentials. Using a Galaxy tool: Database generation The first step in the analysis is to import the required input data files, which users can download from the Shared Data Libraries. Once imported, these data files will appear on the History pane. [clicK on Shared Data tab, and then clicK on “Data Libraries”. 1 In the list of shared data, clicK on “Metaproteomics Training”. ClicK “Metagenome_sixgill.fastq”, select the file, and clicK on “Import to History”. This folder contains one file in the fastq format, which consists of the biological sequence and its corresponding quality scores data. Sixgill (Six-frame Genome-Inferred Libraries for LC-MS/MS) is a tool for using shotgun metagenomics sequencing reads to construct databases of 'metapeptides': short protein fragments for database search of LC-MS/MS metaproteomics data. The main Sixgill command is sixgill_build (http://noble.gs.washington.edu/proj/metapeptide/), which builds a metapeptide database from shotgun metagenomic sequencing reads. -

Next Generation Sequencing Based Forward Genetic Approaches for Identification and Mapping of Causal Mutations in Crop Plants: a Comprehensive Review
plants Review Next Generation Sequencing Based Forward Genetic Approaches for Identification and Mapping of Causal Mutations in Crop Plants: A Comprehensive Review 1, 1, 2,3 2,3 Parmeshwar K. Sahu y , Richa Sao y, Suvendu Mondal , Gautam Vishwakarma , Sudhir Kumar Gupta 2,3, Vinay Kumar 4, Sudhir Singh 2 , Deepak Sharma 1,* and Bikram K. Das 2,3,* 1 Department of Genetics and Plant Breeding, Indira Gandhi Krishi Vishwavidyalaya, Raipur 492012, Chhattisgarh, India; [email protected] (P.K.S.); [email protected] (R.S.) 2 Nuclear Agriculture and Biotechnology Division, Bhabha Atomic Research Centre, Mumbai 400085, India; [email protected] (S.M.); [email protected] (G.V.); [email protected] (S.K.G.); [email protected] (S.S.) 3 Homi Bhabha National Institute, Training School Complex, Anushaktinagar, Mumbai 400094, India 4 ICAR-National Institute of Biotic Stress Management, Baronda, Raipur 493225, Chhattisgarh, India; [email protected] * Correspondence: [email protected] (D.S.); [email protected] (B.K.D.) These authors have contributed equally. y Received: 30 July 2020; Accepted: 21 September 2020; Published: 14 October 2020 Abstract: The recent advancements in forward genetics have expanded the applications of mutation techniques in advanced genetics and genomics, ahead of direct use in breeding programs. The advent of next-generation sequencing (NGS) has enabled easy identification and mapping of causal mutations within a short period and at relatively low cost. Identifying the genetic mutations and genes that underlie phenotypic changes is essential for understanding a wide variety of biological functions. To accelerate the mutation mapping for crop improvement, several high-throughput and novel NGS based forward genetic approaches have been developed and applied in various crops. -

Downloads the Java Archive of Tools
Takeuchi et al. Source Code for Biology and Medicine (2016) 11:12 DOI 10.1186/s13029-016-0058-6 SOFTWARE Open Access cljam: a library for handling DNA sequence alignment/map (SAM) with parallel processing Toshiki Takeuchi* , Atsuo Yamada, Takashi Aoki and Kunihiro Nishimura Abstract Background: Next-generation sequencing can determine DNA bases and the results of sequence alignments are generally stored in files in the Sequence Alignment/Map (SAM) format and the compressed binary version (BAM) of it. SAMtools is a typical tool for dealing with files in the SAM/BAM format. SAMtools has various functions, including detection of variants, visualization of alignments, indexing, extraction of parts of the data and loci, and conversion of file formats. It is written in C and can execute fast. However, SAMtools requires an additional implementation to be used in parallel with, for example, OpenMP (Open Multi-Processing) libraries. For the accumulation of next-generation sequencing data, a simple parallelization program, which can support cloud and PC cluster environments, is required. Results: We have developed cljam using the Clojure programming language, which simplifies parallel programming, to handle SAM/BAM data. Cljam can run in a Java runtime environment (e.g., Windows, Linux, Mac OS X) with Clojure. Conclusions: Cljam can process and analyze SAM/BAM files in parallel and at high speed. The execution time with cljam is almost the same as with SAMtools. The cljam code is written in Clojure and has fewer lines than other similar tools. Keywords: Next-generation sequencing, DNA, Parallel processing, Clojure, SAM, BAM Abbreviations: BAI, BAM index; BGZF, Blocked GNU zip format; LOC, Lines of code; NGS, Next generation sequencing; SAM, Sequence alignment/map Background mainly in FASTQ format, which is a text-based format Next-generation sequencing (NGS) technologies have for sequences and their quality scores.