(Helianthus Annuus L.): Wild Diversity and the Dynamics of Domestication
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Scientific Name Species Common Name Abies Lasiocarpa FIR Subalpine Acacia Macracantha ACACIA Long-Spine
Scientific Name Species Common Name Abies lasiocarpa FIR Subalpine Acacia macracantha ACACIA Long-spine Acacia roemeriana CATCLAW Roemer Acer grandidentatum MAPLE Canyon Acer nigrum MAPLE Black Acer platanoides MAPLE Norway Acer saccharinum MAPLE Silver Aesculus pavia BUCKEYE Red Aesculus sylvatica BUCKEYE Painted Ailanthus altissima AILANTHUS Tree-of-heaven Albizia julibrissin SILKTREE Mimosa Albizia lebbek LEBBEK Lebbek Alnus iridis ssp. sinuata ALDER Sitka Alnus maritima ALDER Seaside Alvaradoa amorphoides ALVARADOA Mexican Amelanchier laevis SERVICEBERRY Allegheny Amyris balsamifera TORCHWOOD Balsam Annona squamosa SUGAR-APPLE NA Araucaria cunninghamii ARAUCARIA Cunningham Arctostaphylos glauca MANZANITA Bigberry Asimina obovata PAWPAW Bigflower Bourreria radula STRONGBACK Rough Brasiliopuntia brasiliensis PRICKLY-PEAR Brazilian Bursera simaruba GUMBO-LIMBO NA Caesalpinia pulcherrima FLOWERFENCE NA Capparis flexuosa CAPERTREE Limber CRUCIFIXION- Castela emoryi THORN NA Casuarina equisetifolia CASUARINA Horsetail Ceanothus arboreus CEANOTHUS Feltleaf Ceanothus spinosus CEANOTHUS Greenbark Celtis lindheimeri HACKBERRY Lindheimer Celtis occidentalis HACKBERRY Common Cephalanthus occidentalis BUTTONBUSH Common Cercis canadensis REDBUD Eastern Cercocarpus traskiae CERCOCARPUS Catalina Chrysophyllum oliviforme SATINLEAF NA Citharexylum berlandieri FIDDLEWOOD Berlandier Citrus aurantifolia LIME NA Citrus sinensis ORANGE Orange Coccoloba uvifera SEAGRAPE NA Colubrina arborescens COLUBRINA Coffee Colubrina cubensis COLUBRINA Cuba Condalia globosa -

Dry White Pine (Hemlock) Oak Forest
Dry white pine (hemlock) oak forest This community type occurs on fairly dry sites, often with 25% or more of the forest floor covered by rocks, boulders and/or exposed bedrock. The canopy may be somewhat open and tree growth somewhat suppressed. The tree stratum is dominated by a mixture of Pinus strobus (eastern white pine), or occasionally Tsuga canadensis (eastern hemlock), and a mixture of dry-site hardwoods, predominantly oaks. On most sites, the conifer and the hardwood component both range between 25% and 75% of the canopy. The oak species most often associated with this type are Quercus montana (chestnut oak), and Q. alba (white oak), although Q. velutina (black oak), Q. coccinea (scarlet oak), or Q. rubra (northern red oak) may also occur. Other associated trees include Nyssa sylvatica (black-gum), Betula lenta (sweet birch), Fraxinus americana (white ash), Prunus serotina (wild black cherry), and Castanea dentata (American chestnut) sprouts. There is often a heath-dominated shrub layer with Kalmia latifolia (mountain laurel) being especially important; Gaylussacia baccata (black huckleberry), Vaccinium spp. (blueberries), and Kalmia angustifolia (sheep laurel) are also common. Other shrubs, like Cornus florida (flowering dogwood), Hamamelis virginiana (witch-hazel), Viburnum acerifolium (maple-leaved viburnum) may also occur on less acidic sites. There is typically a sparse herbaceous layer with a northern affinity; Aralia nudicaulis (wild sarsaparilla), Pteridium aquilinum (bracken fern), Maianthemum canadense (Canada mayflower), Gaultheria procumbens (teaberry), Trientalis borealis (star-flower), and Medeola virginiana (Indian cumber-root) are typical. The successional status of this type seems variable, in some cases, especially on harsher sites, it appears relatively stable, in other cases it appears to be transitional. -

Specializing in Rare and Unique Trees 2015 Catalogue
Whistling Gardens 698 Concession 3, Wilsonville, ON N0E 1Z0 Phone- 519-443-5773 Fax- 519-443-4141 Specializing in Rare and Unique Trees 2015 Catalogue Pot sizes: The number represents the size of the pot ie. #1= 1 gallon, #10 = 10 gallon #1 potted conifers are usually 3-5years old. #10 potted conifers dwarf conifers are between 10 and 15 years old #1 trees= usually seedlings #10 trees= can be several years old anywhere from 5 to 10' tall depending on species and variety. Please ask us on sizes and varieties you are not sure about. Many plants are limited to 1specimen. To reserve your plant(s) a 25% is required. Plants should be picked up by June 15th. Most plants arrive at the gardens by May 10th. Guarantee: We cannot control the weather (good or bad), rodents (big or small), pests (teenie, tiny), poor siting, soil types, lawnmovers, snowplows etc. Plants we carry are expected to grow within the parameters of normal weather conditons. All woody plant purchases are guaranteed from time of purchase to December 1st of current year Any plant not performing or dying in current season will be happily replaced or credited towards a new plant. Please email us if possible with any info needed about our plants. We do not have a phone in the garden centre and I'm rarely in the office. It is very helpful to copy and paste the botanical name of the plant into your Google browser, in most cases, a d Order Item Size Price Description of Pot European White Fir Zone 5 Abies alba Barabit's Spreader 25-30cm #3 $100.00 Abies alba Barabit's Spreader 40cm -

Bioinfo 11 Phylogenetictree
BIO 390/CSCI 390/MATH 390 Bioinformatics II Programming Lecture 11 Phylogenetic Tree Instructor: Lei Qian Fisk University Phylogenetic Tree Applications • Study ancestor-descendant relationships (Evolutionary biology, adaption, genetic drift, selection, speciation, etc.) • Paleogenomics: inferring ancestral genomic information from extinct species (Comparing Chimpanzee, Neanderthal and Human DNA) • Origins of epidemics (Comparing, at the molecular level, various virus strains) • Drug design: specially targeting groups of organisms (Efficient enumeration of phylogenetically informative substrings) • Forensic (Relationships among HIV strains) • Linguistics (Languages tree divergence times) Phylogenetic Tree Illustrating success stories in phylogenetics (I) For roughly 100 years (more exactly, 1870-1985), scientists were unable to figure out which family the giant panda belongs to. Giant pandas look like bears, but have features that are unusual for bears but typical to raccoons: they do not hibernate, they do not roar, their male genitalia are small and backward-pointing. Anatomical features were the dominant criteria used to derive evolutionary relationships between species since Darwin till early 1960s. The evolutionary relationships derived from these relatively subjective observations were often inconclusive. Some of them were later proved incorrect. In 1985, Steven O’Brien and colleagues solved the giant panda classification problem using DNA sequences and phylogenetic algorithms. Phylogenetic Tree Phylogenetic Tree Illustrating success stories in phylogenetics (II) In 1994, a woman from Lafayette, Louisiana (USA), clamed that her ex-lover (who was a physician) injected her with HIV+ blood. Records showed that the physician had drawn blood from a HIV+ patient that day. But how to prove that the blood from that HIV+ patient ended up in the woman? Phylogenetic Tree HIV has a high mutation rate, which can be used to trace paths of transmission. -

Deer-Resistant Landscape Plants This List of Deer-Resistant Landscape Plants Was Compiled from a Variety of Sources
Deer-Resistant Landscape Plants This list of deer-resistant landscape plants was compiled from a variety of sources. The definition of a deer-resistant plant is one that may be occasionally browsed, but not devoured. If deer are hungry enough or there’s a limited amount of food available, they will eat almost anything. Perennials, Herbs & Bulbs Achillea Yarrow Lavandula augustifolia Lavender Adiatum pedatum Maiden Hair Fern Liatris Gayfeather Agastache cana Agastache Lychnis chalcedonica Maltese Cross Ajuga reptans Bugleweed Matteuccia Ostrich Fern Alchemilla Lady’s Mantle Mentha Mint Allium spp. Flowering Onion Miscanthus Silver Grass Arabis Rock Cress Myosotis sylvatica Forget-Me-Not Armeria maritime Sea Thrift Narcissus Daffodils & Narcissus Artemisia Wormwood Nepeta Catmint Asarum europaeum Wild Ginger Oenothera Evening Primrose Asclepias tuberosa Butterfly Weed Paeonia Peony Bergenia Bergenia Papaver orientale Oriental Poppy Calamagrostis Reed Grass Pennisetum orientale Oriental Fountain Grass Cerastium Snow-In-Summer Perovskia Russian Sage Cimcifuga Bugbane Physostegia Obedient Plant Convallaria Lily-of-the-Valley Platycodon Balloon Flower Corydalis lutea Gold Bleeding Heart Polygonatum Solomon’s Seal Dicentra Bleeding Heart Pulmonaria Lungwort Digitalis Foxglove Rudbeckia Black Eyed Susan Echinops Globe Thistle Saponaria Soapwort Festuca ovinia Blue Fescue Salvia Sage Galanthus nivalis Snowdrop Sedum Stonecrop Helleborus Hellebores Solidago Goldenrod Iris sibirica & ensata Japanese & Siberian Iris Stachys byzantina Lamb’s Ear Lamium -
Trees & Conifers
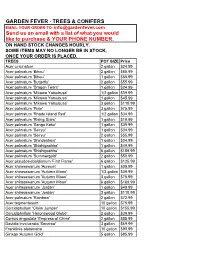
GARDEN FEVER - TREES & CONIFERS EMAIL YOUR ORDER TO: [email protected] Send us an email with a list of what you would like to purchase & YOUR PHONE NUMBER. ON HAND STOCK CHANGES HOURLY. SOME ITEMS MAY NO LONGER BE IN STOCK, ONCE YOUR ORDER IS PLACED. TREES POT SIZE Price Acer ciricinatum 2 gallon $24.99 Acer palmatum 'Bihou' 2 gallon $85.99 Acer palmatum 'Bihou' 1 gallon $55.99 Acer palmatum 'Butterfly' 2 gallon $55.99 Acer palmatum 'Dragon Tears' 1 gallon $24.99 Acer palmatum 'Mikawa Yatsubusa' 1/2 gallon $39.99 Acer palmatum 'Mikawa Yatsubusa' 1 gallon $45.99 Acer palmatum 'Mikawa Yatsubusa' 3 gallon $110.99 Acer palmatum 'Pixie' 3 gallon $75.99 Acer palmatum 'Rhode Island Red' 1/2 gallon $34.99 Acer palmatum 'Rising Stars' 1 gallon $19.99 Acer palmatum 'Sango Kaku' 1 gallon $39.99 Acer palmatum 'Seiryu' 1 gallon $34.99 Acer palmatum 'Seiryu' 2 gallon $55.99 Acer palmatum 'Shindeshojo' 1 gallon $34.99 Acer palmatum 'Shishigashira' 1 gallon $49.99 Acer palmatum 'Shishigashira' 6 gallon $159.99 Acer palmatum 'Summergold' 2 gallon $59.99 Acer pseudosieboldianum 'First Flame' 6 gallon $125.99 Acer shirasawanum 'Aureum' 1 gallon $39.99 Acer shirasawanum 'Autumn Moon' 1/2 gallon $39.99 Acer shirasawanum 'Autumn Moon' 3 gallon $75.99 Acer shirasawanum 'Autumn Moon' 6 gallon $169.99 Acer shirasawanum 'Jordan' 1 gallon $49.99 Acer shirasawanum 'Jordan' 3 gallon $110.99 Acer palmatum 'Rainbow' 2 gallon $72.99 Acer tegmentosum 10 gallon $75.99 Cercidiphyllum 'Claim Jumper' 10 gallon $155.99 Cercidiphyllum 'Heronswood Globe' 2 gallon $39.99 Cornus -

Nursery Catalog
Tel: 503.628.8685 Fax: 503.628.1426 www.eshraghinursery.com 1 Eshraghi’s TOP 10 picks Our locations 1 Main Office, Shipping & Growing 2 Retail Store & Growing 26985 SW Farmington Road Farmington Gardens Hillsboro, OR 97123 21815 SW Farmington Road Beaverton, OR 97007 1 2 3 7 6 3 River Ranch Facility 4 Liberty Farm 4 5 10 N SUNSET HWY TO PORTLAND 8 9 TU HILLSBORO ALA TIN 26 VALL SW 185TH AVE. EY HWY. #4 8 BEAVERTON TONGUE LN. GRABEL RD . D R . E D G R ID E ALOHA R G B D I R R #3 SW 209TH E B T D FARMINGTON ROAD D N A I SIMPSON O O M O R R 10 217 ROSEDALE W R E S W V S I R N W O 219 T K C A J #2 #1 SW UNGER RD. SW 185TH AVE. 1 Acer circinatum ‘Pacific Fire’ (Vine Maple), page 6 D A SW MURRAY BLVD. N RO 2 palmatum (Japanese Maple), NGTO Acer 'Geisha Gone Wild' page 8 FARMI 3 Acer palmatum 'Mikawa yatsubusa' (Japanese Maple), page 10 #1 4 Acer palmatum dissectum 'Orangeola' (Japanese Maple), page 14 5 Hydrangea macrophylla 'McKay', Cherry Explosion PP28757 (Hydrangea), page 32 6 Picea glauca 'Eshraghi1', Poco Verde (White Spruce), page 61 ROAD HILL CLARK 7 Picea pungens 'Hockersmith', Linda (Colorado Spruce), page 64 RY ROAD 8 Pinus nigra 'Green Tower' (Austrian Pine), page 65 SCHOLLS FER 9 Thuja occidentalis 'Janed Gold', Highlights™ PP21967 (Arborvitae), page 70 10 Thuja occidentalis 'Anniek', Sienna Sunset™ (Arborvitae), page 69 Table of contents Tags Make a Difference . -

Hidden Lake Gardens Michigan State University 2019 Plant Sale Plant
Hidden Lake Gardens Michigan State University 2019 Plant Sale Plant List **Please Note: All plants listed are subject to availability due to weather conditions, crop failure, or supplier delivery** Vegetables and Herbs Many assorted varieties Annuals Assorted hanging baskets – Proven Winners® Selections Senecio ‘Angel Wings’ Supertunia® ‘Bordeaux’™ Supertunia® ‘Piccaso in Purple’® Supertunia Royale® Plum Wine Gerbera ‘Garvinea Sweet Sunset’ Salvia ‘Black and Bloom’ Coleus ‘Golden Gate’ Stained Glassworks™ Coleus ‘Royalty’ Stained Glassworks™ Coleus ‘Eruption’ Stained Glassworks™ Coleus ‘Luminesce’ Stained Glassworks™ Coleus ‘Great Falls Niagara’ Strobilanthes dyeriana Persian Shield Bidens ferulifolia ‘Bee Alive’ Bidens ferulifolia ‘Bee Happy’ Bidens ferulifolia ‘Sun Drop Compact’ Zinnia Zahara™ Lantana Lucky™ Angelonia angustifolia Archangel™ Portulaca ColorBlast™ ‘Watermelon Punch’ Perennials Achillea ‘Pomegranate’ Aconitum arendsii Agastache ‘Kudos Coral’ Agastache ‘Kudos Mandarin’ Aquilegia ‘Clementine Dark Purple’ Aquilegia ‘Clementine Salmon Rose’ Armeria maritima ‘Splendens’ Asclepias tuberosa Astilbe ‘Montgomery’ Astilbe ‘Visions’ (purple) Baptisia ‘Lactea’ Baptisia ‘Pink Lemonade’ Bergenia ‘Bressingham Ruby’ Bergenia ‘Sakura’ Brunnera macrophylla ‘Alexander’s Great’ Brunnera macrophylla ‘Jack Frost’ Brunnera macrophylla ‘Sea Heart’ Centaurea montana Ceratostigma plumbaginoides Chelone ‘Hot Lips’ Coreopsis ‘American Dream’ Coreopsis ‘Big Bang Galaxy’ Crocosmia ‘Lucifer’ Dianthus ‘Firewitch’ Dianthus x ‘Kahori’ Dicentra ‘Fire Island’ -

Hemidactylium Scutatum): out of Appalachia and Into the Glacial Aftermath
RANGE-WIDE PHYLOGEOGRAPHY OF THE FOUR-TOED SALAMANDER (HEMIDACTYLIUM SCUTATUM): OUT OF APPALACHIA AND INTO THE GLACIAL AFTERMATH Timothy A. Herman A Thesis Submitted to the Graduate College of Bowling Green State University in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE August 2009 Committee: Juan Bouzat, Advisor Christopher Phillips Karen Root ii ABSTRACT Juan Bouzat, Advisor Due to its limited vagility, deep ancestry, and broad distribution, the four-toed salamander (Hemidactylium scutatum) is well suited to track biogeographic patterns across eastern North America. The range of the monotypic genus Hemidactylium is highly disjunct in its southern and western portions, and even within contiguous portions is highly localized around pockets of preferred nesting habitat. Over 330 Hemidactylium genetic samples from 79 field locations were collected and analyzed via mtDNA sequencing of the cytochrome oxidase 1 gene (co1). Phylogenetic analyses showed deep divergences at this marker (>10% between some haplotypes) and strong support for regional monophyletic clades with minimal overlap. Patterns of haplotype distribution suggest major river drainages, both ancient and modern, as boundaries to dispersal. Two distinct allopatric clades account for all sampling sites within glaciated areas of North America yet show differing patterns of recolonization. High levels of haplotype diversity were detected in the southern Appalachians, with several members of widely ranging clades represented in the region as well as other unique, endemic, and highly divergent lineages. Bayesian divergence time analyses estimated the common ancestor of all living Hemidactylium included in the study at roughly 8 million years ago, with the most basal splits in the species confined to the Blue Ridge Mountains. -

Molecular Phylogenetics and Evolution 55 (2010) 153–167
Molecular Phylogenetics and Evolution 55 (2010) 153–167 Contents lists available at ScienceDirect Molecular Phylogenetics and Evolution journal homepage: www.elsevier.com/locate/ympev Conservation phylogenetics of helodermatid lizards using multiple molecular markers and a supertree approach Michael E. Douglas a,*, Marlis R. Douglas a, Gordon W. Schuett b, Daniel D. Beck c, Brian K. Sullivan d a Illinois Natural History Survey, Institute for Natural Resource Sustainability, University of Illinois, Champaign, IL 61820, USA b Department of Biology and Center for Behavioral Neuroscience, Georgia State University, Atlanta, GA 30303-3088, USA c Department of Biological Sciences, Central Washington University, Ellensburg, WA 98926, USA d Division of Mathematics & Natural Sciences, Arizona State University, Phoenix, AZ 85069, USA article info abstract Article history: We analyzed both mitochondrial (MT-) and nuclear (N) DNAs in a conservation phylogenetic framework to Received 30 June 2009 examine deep and shallow histories of the Beaded Lizard (Heloderma horridum) and Gila Monster (H. Revised 6 December 2009 suspectum) throughout their geographic ranges in North and Central America. Both MTDNA and intron Accepted 7 December 2009 markers clearly partitioned each species. One intron and MTDNA further subdivided H. horridum into its Available online 16 December 2009 four recognized subspecies (H. n. alvarezi, charlesbogerti, exasperatum, and horridum). However, the two subspecies of H. suspectum (H. s. suspectum and H. s. cinctum) were undefined. A supertree approach sus- Keywords: tained these relationships. Overall, the Helodermatidae is reaffirmed as an ancient and conserved group. Anguimorpha Its most recent common ancestor (MRCA) was Lower Eocene [35.4 million years ago (mya)], with a 25 ATPase Enolase my period of stasis before the MRCA of H. -

Native Trees of Massachusetts
Native Trees of Massachusetts Common Name Latin Name Eastern White Pine Pinus strobus Common Name Latin Name Mountain Pine Pinus mugo Pin Oak Quercus palustris Pitch Pine Pinus rigida White Oak Quercus alba Red Pine Pinus resinosa Swamp White Oak Quercus bicolor Scots Pine Pinus sylvestris Chestnut Oak Quercus montana Jack Pine Pinus banksiana Eastern Cottonwood Populus deltoides Eastern Hemlock Tsuga canadensis https://plants.usda.gov/ Tamarack Larix laricina core/profile?symbol=P Black Spruce Picea mariana ODE3 White Spruce Picea glauca black willow Salix nigra Red Spruce Picea rubens Red Mulberry Morus rubra Norway Spruce Picea abies American Plum Prunus americana Northern White cedar Thuja occidentalis Canada Plum Prunus nigra Eastern Juniper Juniperus virginiana Black Cherry Prunus serotina Balsam Fir Abies balsamea Canadian Amelanchier canadensis American Sycamore Platanus occidentalis Serviceberry or Witchhazel Hamamelis virginiana Shadbush Honey Locust Gleditsia triacanthos American Mountain Sorbus americana Eastern Redbud Cercis canadensis Ash Yellow-Wood Cladrastis kentukea American Elm Ulmus americana Gray Birch Betula populifolia Slippery Elm Ulmus rubra Grey Alder Alnus incana Basswood Tilia americana Sweet Birch Betula lenta Smooth Sumac Rhus glabra Yellow Birch Betula alleghaniensis Red Maple Acer rubrum Heartleaf Paper Birch Betula cordifolia Horse-Chestnut Aesculus hippocastanum River Birch Betula nigra Staghorn Sumac Rhus typhina Smooth Alder Alnus serrulata Silver Maple Acer saccharinum American Ostrya virginiana Sugar Maple Acer saccharum Hophornbeam Boxelder Acer negundo American Hornbeam Carpinus caroliniana Black Tupelo Nyssa sylvatica Green Alder Alnus viridis Flowering Dogwood Cornus florida Beaked Hazelnut Corylus cornuta Northern Catalpa Catalpa speciosa American Beech Fagus grandifolia Black Ash Fraxinus nigra Black Oak Quercus velutina Devil's Walkingstick Aralia spinosa Downy Hawthorn Crataegus mollis. -

Chestnut Oak Forest (White Pine Subtype)
CHESTNUT OAK FOREST (WHITE PINE SUBTYPE) Concept: The White Pine Subtype encompasses lower elevation Quercus montana forests that have a significant component of Pinus strobus, which may range from a substantial minority to codominant in the canopy. The lower strata in this subtype appear to overlap the less extreme range of variation of the Dry Heath Subtype and the Herb Subtype. Distinguishing Features: Chestnut Oak Forest (White Pine Subtype) is distinguished from all other communities by the combination of Quercus montana with Pinus strobus, without a component of Quercus alba. The White Pine Subtype subtype should only be used where white pine is believed to be naturally present, not for forests where it has been planted or where it likely spread from nearby plantings. Forests with a more mesophytic composition, such as the forests of Quercus rubra and Pinus strobus with Rhododendron maximum that occur around Linville Falls, are treated as the Mesic Subtype. Synonyms: Pinus strobus - Quercus (coccinea, prinus) / (Gaylussacia ursina, Vaccinium stamineum) Forest (CEGL007519). Ecological Systems: Southern Appalachian Oak Forest (CES202.886). Sites: The White Pine Subtype occurs on open slopes and spur ridges. It often is in steep gorges or in the most rugged foothill ranges. Most example are at 1000-2500 feet elevation, but a few are reported upward to 4000 feet or even higher. Soils: Soils are generally Typic Dystrochrepts, especially Chestnut and Edneyville, less often Typic Hapludults such as Tate or Cowee. Hydrology: Sites are dry and well drained but may be somewhat less stressful because of topographic sheltering. Vegetation: The forest canopy is dominated by a mix of Pinus strobus and Quercus montana, occasionally with Quercus coccinea or Quercus rubra codominant.