Long Term Blood Oxygenation Membranes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Properties of Polypropylene Yarns with a Polytetrafluoroethylene
coatings Article Properties of Polypropylene Yarns with a Polytetrafluoroethylene Coating Containing Stabilized Magnetite Particles Natalia Prorokova 1,2,* and Svetlana Vavilova 1 1 G.A. Krestov Institute of Solution Chemistry of the Russian Academy of Sciences, Akademicheskaya St. 1, 153045 Ivanovo, Russia; [email protected] 2 Department of Natural Sciences and Technosphere Safety, Ivanovo State Polytechnic University, Sheremetevsky Ave. 21, 153000 Ivanovo, Russia * Correspondence: [email protected] Abstract: This paper describes an original method for forming a stable coating on a polypropylene yarn. The use of this method provides this yarn with barrier antimicrobial properties, reducing its electrical resistance, increasing its strength, and achieving extremely high chemical resistance, similar to that of fluoropolymer yarns. The method is applied at the melt-spinning stage of polypropylene yarns. It is based on forming an ultrathin, continuous, and uniform coating on the surface of each of the yarn filaments. The coating is formed from polytetrafluoroethylene doped with magnetite nanoparticles stabilized with sodium stearate. The paper presents the results of a study of the effects of such an ultrathin polytetrafluoroethylene coating containing stabilized magnetite particles on the mechanical and electrophysical characteristics of the polypropylene yarn and its barrier antimicrobial properties. It also evaluates the chemical resistance of the polypropylene yarn with a coating based on polytetrafluoroethylene doped with magnetite nanoparticles. Citation: Prorokova, N.; Vavilova, S. Properties of Polypropylene Yarns Keywords: coatings; polypropylene yarn; polytetrafluoroethylene; magnetite nanoparticles; barrier with a Polytetrafluoroethylene antimicrobial properties; surface electrical resistance; chemical resistance; tensile strength Coating Containing Stabilized Magnetite Particles. Coatings 2021, 11, 830. https://doi.org/10.3390/ coatings11070830 1. -

Chlorine Dioxide Compatibility Assessment
Chlorine Dioxide Compatibility Assessment DRF3366 - TECH-COMM-001 Created by: Tristel Solutions Limited, Lynx Business Park, Cambs, UK, CB8 7NY Copyright © Tristel Solutions T +44 (0) 1638 721500 – E [email protected] – W www.tristel.com January 2018 Key - Excellent - no change at all in the device - Good - slight cosmetic change/ decolourisation – material/device still fully functional - Fair – slight damage is observed– material/device still fully functional - Poor – visible pitting, breaking, brittle METALS Materials of construction 1 2 3 4 (Excellent) (Good) (Fair) (Poor) Stainless Steel 317 (Pure) X Stainless Steel 316 (pure, fully X austenitic with no ferric content) Stainless Steel 304 X (High grade, low ferric content) Hastelloy X Titanium X Aluminium X Aluminium (Anodised) X Brass X Copper X Chrome X Mild Steel X DRF3366 - TECH-COMM-001 Created by: Tristel Solutions Limited, Lynx Business Park, Cambs, UK, CB8 7NY Copyright © Tristel Solutions T +44 (0) 1638 721500 – E [email protected] – W www.tristel.com January 2018 PLASTICS Materials of construction 1 2 3 4 (Excellent) (Good) (Fair) (Poor) Perfluoroelastomer X PEEK X X Polypropylene X PVDF X X Polycarbonate X Polysulphone (natural) X Polysulphone (white) X PET-P X PMMA X Polyamide X Polyethylene X Polyoxymethylene X Acetal X ABS X Polymethylpentene (TPX) (PMP) X DRF3366 - TECH-COMM-001 Created by: Tristel Solutions Limited, Lynx Business Park, Cambs, UK, CB8 7NY Copyright © Tristel Solutions T +44 (0) 1638 721500 – E [email protected] – W www.tristel.com January 2018 PLASTICS -

POLYPROPYLENE Chemical Resistance Guide
POLYPROPYLENE Chemical Resistance Guide SECOND EDITION PP CHEMICAL RESISTANCE GUIDE Thermoplastics: Polypropylene (PP) Chemical Resistance Guide Polypropylene (PP) 2nd Edition © 2020 by IPEX. All rights reserved. No part of this book may be used or reproduced in any manner whatsoever without prior written permission. For information contact: IPEX, Marketing, 1425 North Service Road East, Oakville, Ontario, Canada, L6H 1A7 About IPEX At IPEX, we have been manufacturing non-metallic pipe and fittings since 1951. We formulate our own compounds and maintain strict quality control during production. Our products are made available for customers thanks to a network of regional stocking locations from coast-to-coast. We offer a wide variety of systems including complete lines of piping, fittings, valves and custom-fabricated items. More importantly, we are committed to meeting our customers’ needs. As a leader in the plastic piping industry, IPEX continually develops new products, modernizes manufacturing facilities and acquires innovative process technology. In addition, our staff take pride in their work, making available to customers their extensive thermoplastic knowledge and field experience. IPEX personnel are committed to improving the safety, reliability and performance of thermoplastic materials. We are involved in several standards committees and are members of and/or comply with the organizations listed on this page. For specific details about any IPEX product, contact our customer service department. xx: Max Recommended Temperature – Unsuitable / Insufficient Data A: Applicable in Some Cases, consult IPEX 2 IPEX Chemical Resistance Guide for PP INTRODUCTION Thermoplastics and elastomers have outstanding resistance to a wide range of chemical reagents. The chemical resistance of plastic piping is basically a function of the thermoplastic material and the compounding components. -

Development, Structure and Strength Properties of PP/PMMA/FA Blends
Bull. Mater. Sci., Vol. 23, No. 2, April 2000, pp. 103–107. © Indian Academy of Sciences. Development, structure and strength properties of PP/PMMA/FA blends NAVIN CHAND* and S R VASHISHTHA Regional Research Laboratory, Habibganj Naka, Bhopal 462 026, India MS received 10 September 1999; revised 29 January 2000 Abstract. A new type of flyash filled PP/PMMA blend has been developed. Structural and thermal properties of flyash (FA) filled polypropylene (PP)/polymethyl methacrylate (PMMA) blend system have been determined and analysed. Filled polymer blends were developed on a single screw extruder. Strength and thermal pro- perties of FA filled and unfilled PP/PMMA blends were determined. Addition of flyash imparted dimensional and thermal stability, which has been observed in scanning electron micrographs and in TGA plot. Increase of flyash concentration increased the initial degradation temperature of PP/PMMA blend. The increase of thermal stability has been explained based on increased mechanical interlocking of PP/PMMA chains inside the hollow structure of flyash. Keywords. Flyash polymer composite; morphology; particulate. 1. Introduction polarity and the polymer with polar groups facilitate sur- face bonding because the surface of filler can be easily Polypropylene (PP) is a commercially important polymer, wetted by a polymer. Therefore, whether or not the poly- which is of practical use in a wide range of applications mer molecule has polarity is an important parameter in (Glenz 1986). Its morphology (Frank 1968) means that determining the reinforcement effect of polymer compo- the mechanical properties of PP are moderate, so if one sites. Chemical bonding between polymer matrix and wants to extend the field of application of this material, an filler offers good bonding of constituents. -

Wood Fiber Reinforcement of Styrene-Maleic Anhydride Copolymers
The Fourth International Conference on Woodfiber-Plastic Composites Wood Fiber Reinforcement of Styrene-Maleic Anhydride Copolymers John Simonsen Rodney Jacobson Roger Rowell Abstract Styrene-maleic anhydride (SMA) copolymers are We also investigated the thermal properties of the used in the automotive industry, primarily for interior composites using differential scanning calorimetry. parts. The use of wood fillers may provide a more While the SMA molecule contains an anhydride economical means of manufacturing these items group, we observed no evidence of chemical bonding and/or improve their properties. We made a prelimi- between SMA and the wood filler. nary study of the feasibility of utilizing wood-based Introduction fillers in SMA copolymers, comparing the mechanical The development of useful wood-filled thermo- properties of wood-filled SMA to wood-filled poly- styrene. The fillers studied were fibers for aspen plastic products has received increasing attention medium density fiberboard, pine wood flour, and from manufacturers and the research community milled recycled newsprint, which showed unusually recently. This is due to the low cost and easy proc- high properties considering its size and aspect ratio. essability of wood fillers. In addition, wood fillers can add reinforcement in some cases, improving the properties of the final composite over those of the unfilled plastic (6, 7, 10–12). Simonsen: Styrene-maleic anhydride (SMA) copolymers are Assistant Professor, Dept- of Forest Prod., Oregon State utilized in the automotive industry for the injection University, Corvallis, Oregon molding and thermoforming of interior parts, The Jacobson: superiority of SMA over polystyrene is due to its Materials Engineer, USDA Forest Serv., Forest Prod. -

Effects of Polypropylene, Polyvinyl Chloride, Polyethylene Terephthalate, Polyurethane, High
Effects of polypropylene, polyvinyl chloride, polyethylene terephthalate, polyurethane, high- density polyethylene, and polystyrene microplastic on Nelumbo nucifera (Lotus) in water and sediment Maranda Esterhuizen ( maranda.esterhuizen@helsinki. ) University of Helsinki: Helsingin Yliopisto https://orcid.org/0000-0002-2342-3941 Youngjun Kim Korea Institute of Science and Technology Europe Forschungsgesellschaft mbH Research Article Keywords: Microplastics, oxidative stress, sediment, macrophyte, exposure, germination, seedling growth Posted Date: May 11th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-458889/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Loading [MathJax]/jax/output/CommonHTML/jax.js Page 1/20 Abstract Plastic waste is recognised as hazardous, with the risk increasing as the polymers break down in nature to secondary microplastics or even nanoplastics. The number of studies reporting on the prevalence of microplastic in every perceivable niche and bioavailable to biota is dramatically increasing. Knowledge of the ecotoxicology of microplastic is advancing as well; however, information regarding plants, specically aquatic macrophytes, is still lacking. The present study aimed to gain more information on the ecotoxicological effects of six different polymer types as 4 mm microplastic on the morphology (germination and growth) and the physiology (catalase and glutathione S-transferase activity) of the rooted aquatic macrophyte, Nelumbo nucifera. The role of sediment was also considered by conducting all exposure both in a sediment-containing and sediment-free exposure system. Polyvinyl chloride and polyurethane exposures caused the highest inhibition of germination and growth compared to the control. However, the presence of sediment signicantly decreased the adverse effects. -

Functionalization of Polyethylene (PE) and Polypropylene (PP) Material Using Chitosan Nanoparticles with Incorporated Resveratrol As Potential Active Packaging
materials Article Functionalization of Polyethylene (PE) and Polypropylene (PP) Material Using Chitosan Nanoparticles with Incorporated Resveratrol as Potential Active Packaging Tjaša Kraševac Glaser 1,*, Olivija Plohl 1, Alenka Vesel 2, Urban Ajdnik 1 , Nataša Poklar Ulrih 3, Maša Knez Hrnˇciˇc 4, Urban Bren 4 and Lidija Fras Zemljiˇc 1,* 1 Laboratory for Characterization and Processing of Polymers, Faculty of Mechanical Engineering, University of Maribor, Smetanova 17, SI-2000 Maribor, Slovenia 2 Department of Surface Engineering and Optoelectronics, Jožef Stefan Institute, Teslova 30, SI-1000 Ljubljana, Slovenia 3 Department of Food Science and Technology, Biotechnical Faculty, University of Ljubljana, Jamnikarjeva 101, SI-1000 Ljubljana, Slovenia 4 Faculty of Chemistry and Chemical Engineering, University of Maribor, Smetanova 17, SI-2000 Maribor, Slovenia * Correspondence: [email protected] (T.K.G.); [email protected] (L.F.Z.); Tel.: +386-2-220-7607 (T.G.K.); +386-2-220-7909 (L.F.Z.) Received: 21 May 2019; Accepted: 28 June 2019; Published: 1 July 2019 Abstract: The present paper reports a novel method to improve the properties of polyethylene (PE) and polypropylene (PP) polymer foils suitable for applications in food packaging. It relates to the adsorption of chitosan-colloidal systems onto untreated and oxygen plasma-treated foil surfaces. It is hypothesized that the first coated layer of chitosan macromolecular solution enables excellent antibacterial properties, while the second (uppermost) layer contains a network of polyphenol resveratrol, embedded into chitosan nanoparticles, which enables antioxidant and antimicrobial properties simultaneously. X-ray photon spectroscopy (XPS) and infrared spectroscopy (FTIR) showed successful binding of both coatings onto foils as confirmed by gravimetric method. -

Bio-Based and Biodegradable Plastics – Facts and Figures Focus on Food Packaging in the Netherlands
Bio-based and biodegradable plastics – Facts and Figures Focus on food packaging in the Netherlands Martien van den Oever, Karin Molenveld, Maarten van der Zee, Harriëtte Bos Rapport nr. 1722 Bio-based and biodegradable plastics - Facts and Figures Focus on food packaging in the Netherlands Martien van den Oever, Karin Molenveld, Maarten van der Zee, Harriëtte Bos Report 1722 Colophon Title Bio-based and biodegradable plastics - Facts and Figures Author(s) Martien van den Oever, Karin Molenveld, Maarten van der Zee, Harriëtte Bos Number Wageningen Food & Biobased Research number 1722 ISBN-number 978-94-6343-121-7 DOI http://dx.doi.org/10.18174/408350 Date of publication April 2017 Version Concept Confidentiality No/yes+date of expiration OPD code OPD code Approved by Christiaan Bolck Review Intern Name reviewer Christaan Bolck Sponsor RVO.nl + Dutch Ministry of Economic Affairs Client RVO.nl + Dutch Ministry of Economic Affairs Wageningen Food & Biobased Research P.O. Box 17 NL-6700 AA Wageningen Tel: +31 (0)317 480 084 E-mail: [email protected] Internet: www.wur.nl/foodandbiobased-research © Wageningen Food & Biobased Research, institute within the legal entity Stichting Wageningen Research All rights reserved. No part of this publication may be reproduced, stored in a retrieval system of any nature, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, without the prior permission of the publisher. The publisher does not accept any liability for inaccuracies in this report. 2 © Wageningen Food & Biobased Research, institute within the legal entity Stichting Wageningen Research Preface For over 25 years Wageningen Food & Biobased Research (WFBR) is involved in research and development of bio-based materials and products. -
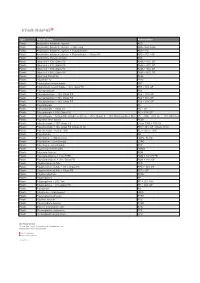
Type Material Name Abbreviation Plastic Acrylonitrile Butadiene
Type Material Name Abbreviation Plastic Acrylonitrile butadiene styrene ABS Plastic Acrylonitrile butadiene styrene - High-Temp ABS - high temp Plastic Acrylonitrile butadiene styrene + Polycarbonate ABS + PC Plastic Acrylonitrile butadiene styrene + Polycarbonate + Glass Fill ABS + PC + GF Plastic Acrylonitrile styrene acrylate ASA Plastic Nylon 6-6 + 10% Glass Fill PA66 + 10% GF Plastic Nylon 6-6 + 20% Glass Fill PA66 + 20% GF Plastic Nylon 6-6 + 30% Glass Fill PA66 + 30% GF Plastic Nylon 6-6 + 50% Glass Fill PA66 + 50% GF Plastic Nylon 6-6 Polyamide PA66 Plastic Polyamide 12 PA12 Plastic Polybutylene terephthalate PBT Plastic Polybutylene terephthalate + 30% Glass Fill PBT+ 30% GF Plastic Polycaprolactam PA6 Plastic Polycaprolactam + 20% Glass Fill PA6 + 20% GF Plastic Polycaprolactam + 30% Glass Fill PA6 + 30% GF Plastic Polycaprolactam + 50% Glass Fill PA6 + 50% GF Plastic Polycarbonate PC Plastic Polycarbonate + Glass Fill PC + GF Plastic Polycarbonate + 10% Glass Fill PC + 10% GF Plastic Polycarbonate + Acrylonitrile butadiene styrene + 20% Glass Fill + 10% Stainless Steel fiber PC + ABS + 20% GF + 10% SS Fiber Plastic Polyether ether ketone PEEK Plastic Polyetherimide + 30% Glass Fill Ultem 1000 + 30% GF Plastic Polyetherimide + 40% Glass Fill (Ultem 2410) PEI + 40% GF (Ultem 2410) Plastic Polyetherimide + Ultem 1000 PEI + Ultem 1000 Plastic Polyethylene PE Plastic Polyethylene - High-Density HDPE, PEHD Plastic Polyethylene - Low-Density LDPE Plastic Polyethylene terephthalate PET Plastic Polymethyl methacrylate PMMA Plastic Polyoxymethylene -

High Performance Waxadditives
High Performance Wax Additives Advanced technology with a quality difference MICRO POWDERS, INC. 580 White Plains Road, Tarrytown, New York 10591 • Tel: (914) 793-4058, Fax: (914) 472-7098 • Email: [email protected] Quality, Innovation and Partnership Support Since 1971 About MPI For reliable quality and superb consistency in wax additives, formulators rely on Micro Powders®, the recognized leader in advanced wax technology. Our specialty products meet the demanding requirements of diverse markets, from paints, coatings and printing inks to ceramics, lubricants, adhesives and more. Our extensive range of micronized waxes, wax dispersions and wax emulsions brings the right solution to a vast array of applications, with reliable batch-to-batch consistency and superior performance values. Ongoing innovation keeps Micro Powders ahead of the curve in responding to industry trends. R&D takes place in our Advanced Applications Lab, staffed by chemists with many years experience in the industries we serve. The Micro Powders quality assurance system is certified to ISO 9001. Along with our technical expertise comes dedicated partnership support from our knowledgeable distributors worldwide and our own staff experts. Our advanced technology will make a quality difference in your products, and your profits. Unique Products Laser Diffraction Analysis ensures consistent particle size uniformity from batch-to-batch. Our wax additives are easily dispersed without prior melting or grinding. Product groups include: MP Synthetic Waxes for lubricity -

TPX® Datasheet
Metals & Materials for Research and Industry TPX® Datasheet GOODFELLOW & MITSUI & CO. EUROPE PLC TPX® Polymethylpentene (PMP) TPX®, a 4-methylpentene-1 based polyolefin manufactured solely by Mitsui Chemicals, Inc. is now available in small order lots from Goodfellow. This lightweight, functional polymer displays a unique combination of physical properties and characteristics due to its distinctive molecular structure. Because of its superb chemical resistance and optical clarity TPX® is an ideal choice when replacing polycarbonate or acrylic in some chemically demanding environments. • High heat resistance (melt temperature 236°C) • Lowest specific gravity of any known polymer (0.833 g/cc) CH CH2 CH CH2 • Superior steam sterilisation capabilities • Superior transparency to visible light CH CH • Excellent UV transmission characteristics 2 2 • Lower refractive index than other transparent resins CH CH • Excellent acoustic properties • Excellent chemical resistance H C CH H C CH • High gas and moisture permeability 3 3 3 3 • Outstanding dielectric properties The properties of TPX® make it ideal for extruding items such as film and paper coating, injection moulded items and blow moulded components, and we list some of the possible applications below: • Release film • Release paper for synthetic leather • LED moulds • Mandrels and sheaths for hose and cable use • Cosmetic caps and tubes • Heat resistant non-woven fabric and fibres • Chemical tubes • Animal cages • Sterilisation containers Goodfellow Cambridge Ltd [email protected] Ermine -

Bioplastics Market Data 2019
REPORT European Bioplastics Bioplastics market data 2019 Global production capacities of bioplastics 2019-2024 1 Global production capacities of bioplastics 2019-2024 Dynamic market growth continues Global production capacities of bioplastics 2019 vs. 2024 Currently, bioplastics represent about one percent of the about 360 million tonnes* of plastic produced annually. But, as demand is rising, and with more sophisticated biopoly- mers, applications, and products emerging, the market is continuously growing. According to the latest market data compiled by European Bioplastics in cooperation with the research institute nova- Institute, global bioplastics production capacity is set to in- crease from around 2.11 million tonnes in 2019 to approxi- mately 2.43 million tonnes in 2024. Development of innovative materials New and innovative biopolymers, such as bio-based PP (polypropylene) and PHAs (polyhydroxyalkanoates) show the highest relative growth rates. In 2019, bio-based PP en- tered the market at commercial scale with a strong growth potential due to the widespread application of PP in a wide range of sectors. Their production capacities are predicted to almost sextuple by 2024. PHAs are an important polymer family, whose production capacities are estimated to more than triple in the next five years. These polyesters are 100 percent bio-based and biodegradable and feature a wide array of physical and mechanical properties depending on their chemical composition. Global production capacities of bioplastics 2019 (by material type) Bio-based, non-biodegradable plastics altogether, includ- ing also the drop-in solutions bio-based PE (polyethylene) and bio-based PET (polyethylene terephthalate), as well as bio-based PA (polyamides), currently make up for over 44 percent (almost 1 million tonnes) of the global bioplastics production capacities.