Copyright and Citation Considerations for This Thesis/ Dissertation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chemical Process Modeling in Modelica
Chemical Process Modeling in Modelica Ali Baharev Arnold Neumaier Fakultät für Mathematik, Universität Wien Nordbergstraße 15, A-1090 Wien, Austria Abstract model creation involves only high-level operations on a GUI; low-level coding is not required. This is the Chemical process models are highly structured. Infor- desired way of input. Not surprisingly, this is also mation on how the hierarchical components are con- how it is implemented in commercial chemical process nected helps to solve the model efficiently. Our ulti- simulators such as Aspen PlusR , Aspen HYSYSR or mate goal is to develop structure-driven optimization CHEMCAD R . methods for solving nonlinear programming problems Nonlinear system of equations are generally solved (NLP). The structural information retrieved from the using optimization techniques. AMPL (FOURER et al. JModelica environment will play an important role in [12]) is the de facto standard for model representation the development of our novel optimization methods. and exchange in the optimization community. Many Foundations of a Modelica library for general-purpose solvers for solving nonlinear programming (NLP) chemical process modeling have been built. Multi- problems are interfaced with the AMPL environment. ple steady-states in ideal two-product distillation were We are aiming to create a ‘Modelica to AMPL’ con- computed as a proof of concept. The Modelica source verter. One could use the Modelica toolchain to create code is available at the project homepage. The issues the models conveniently on a GUI. After exporting the encountered during modeling may be valuable to the Modelica model in AMPL format, the already existing Modelica language designers. software environments (solvers with AMPL interface, Keywords: separation, distillation column, tearing AMPL scripts) can be used. -

Chemical Engineering Careers in the Bioeconomy
BioFutures Chemical engineering careers in the bioeconomy A selection of career profiles Foreword In December 2018, IChemE published the final report of its BioFutures Programme.1 The report recognised the need for chemical engineers to have a greater diversity of knowledge and skills and to be able to apply these to the grand challenges facing society, as recognised by the UN Sustainable Development Goals2 and the NAE Grand Challenges for Engineering.3 These include the rapid development of the bioeconomy, pressure to reduce greenhouse gas emissions, and an increased emphasis on responsible and sustainable production. One of the recommendations from the BioFutures report prioritised by IChemE’s Board of Trustees was for IChemE to produce and promote new career profiles to showcase the roles of chemical engineers in the bioeconomy, in order to raise awareness of their contribution. It gives me great pleasure to present this collection of careers profiles submitted by members of the chemical engineering community. Each one of these career profiles demonstrates the impact made by chemical engineers across the breadth of the bioeconomy, including water, energy, food, manufacturing, and health and wellbeing. In 2006, the Organisation for Economic Co-operation and Development (OECD) defined the bioeconomy as “the aggregate set of economic operations in a society that uses the latent value incumbent in biological products and processes to capture new growth and welfare benefits for citizens and nations”.4 This definition includes the use of biological feedstocks and/or processes which involve biotechnology to generate economic outputs. The output in terms of products and services may be in the form of chemicals, food, pharmaceuticals, materials or energy. -

Yntbietic Fnei
23c$b/J jflt yntbietic fnei OIL SHALE 0 COAL 0 OIL SANDS VOLUME 24 — NUMBER 3 — SEPTEMBER 1987 QUARTERLY Toll Eril Reposito,y ,.r;.'.ur Lakas Library csdo School of Mcs © THE PACE CONSULTANTS INC. 0 Reg. U.S. Pal. OFF. Pace Synthetic Fuels Report Is published by The Pace Consultants Inc., as a multi-client service and is intended for the sole use of the clients or organizations affiliated with clients by virtue of a relationship equivalent to 51 percent or greater ownership. Pace Synthetic Fuels Report is protected by the copyright laws of the United States; reproduction of any part of the publication requires the express permission of The Pace Con- sultants Inc. The Pace Consultants Inc., has provided energy consulting and engineering services since 1955. The company's experience includes resource evalua- tion, process development and design, systems planning, marketing studies, licenser comparisons, environmental planning, and economic analysis. The Synthetic Fuels Analysis group prepares a variety of periodic and other reports analyzing developments in the energy field. THE PACE CONSULTANTS INC. SYNTHETIC FUELS ANALYSIS MANAGING EDITOR Jerry E. Sinor Pt Office Box 649 Niwot, Colorado 80544 (303) 652-2632 BUSINESS MANAGER Ronald L. Gist Post Office Box 53473 Houston, Texas 77052 (713) 669-8800 Telex: 77-4350 CONTENTS HIGHLIGHTS A-i I. GENERAL CORPORATIONS Gill Cuts Coal Gasification Programs 1-i API Data Shows Drop in Oil Production 1-4 GOVERNMENT Petroleum Research Institute Proposed i-S FERC Kills Incremental Gas Pricing 1-5 California to Fund Advanced Energy Technologies i_s ERAB Studying Energy Competitiveness 1-6 ENERGY POLICY AND FORECASTS U.S. -

Handbook of Chemical Engineering Calculations
Source: HANDBOOK OF CHEMICAL ENGINEERING CALCULATIONS SECTION 1 PHYSICAL AND CHEMICAL PROPERTIES Avinash Gupta, Ph.D. Senior Principal Chemical Engineer Chevron Lummus Global Bloomfield, NJ 1.1 MOLAR GAS CONSTANT 1.2 EQUATIONS AND LIQUID-LIQUID 1.2 ESTIMATION OF CRITICAL EQUILIBRIUM DATA 1.44 TEMPERATURE FROM EMPIRICAL 1.21 ACTIVITY-COEFFICIENT-CORRELATION CORRELATION 1.2 EQUATIONS AND VAPOR-LIQUID 1.3 CRITICAL PROPERTIES FROM EQUILIBRIUM DATA 1.46 GROUP-CONTRIBUTION METHOD 1.3 1.22 CONVERGENCE-PRESSURE 1.4 REDLICH-KWONG EQUATION VAPOR-LIQUID EQUILIBRIUM K OF STATE 1.5 VALUES 1.49 1.5 P-V-T PROPERTIES OF A GAS 1.23 HEAT OF FORMATION FROM MIXTURE 1.8 ELEMENTS 1.64 1.6 DENSITY OF A GAS MIXTURE 1.12 1.24 STANDARD HEAT OF REACTION, 1.7 ESTIMATION OF LIQUID STANDARD FREE-ENERGY CHANGE, AND EQUILIBRIUM CONSTANT 1.67 DENSITY 1.14 1.25 STANDARD HEAT OF REACTION FROM 1.8 ESTIMATION OF IDEAL-GAS HEAT HEAT OF FORMATION—AQUEOUS CAPACITY 1.15 SOLUTIONS 1.68 1.9 HEAT CAPACITY OF REAL GASES 1.20 1.26 STANDARD HEAT OF REACTION FROM 1.10 LIQUID HEAT CAPACITY— HEAT OF COMBUSTION 1.68 GENERALIZED CORRELATION 1.22 1.27 STANDARD HEAT OF FORMATION 1.11 ENTHALPY DIFFERENCE FOR IDEAL FROM HEAT OF COMBUSTION 1.70 GAS 1.24 1.28 HEAT OF ABSORPTION FROM 1.12 ESTIMATION OF HEAT OF SOLUBILITY DATA 1.71 VAPORIZATION 1.24 1.29 ESTIMATION OF LIQUID VISCOSITY 1.13 PREDICTION OF VAPOR AT HIGH TEMPERATURES 1.73 PRESSURE 1.27 1.30 VISCOSITY OF NONPOLAR AND 1.14 ENTHALPY ESTIMATION— POLAR GASES AT HIGH GENERALIZED METHOD 1.29 PRESSURE 1.73 1.15 ENTROPY INVOLVING A PHASE 1.31 -

Green Petroleum Refining - Mathematical Models for Optimizing Petroleum Refining Under Emission Constraints
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by University of Waterloo's Institutional Repository Green Petroleum Refining - Mathematical Models for Optimizing Petroleum Refining Under Emission Constraints by Yusuf Ali Yusuf A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree of Master of Applied Science in Chemical Engineering Waterloo, Ontario, Canada, 2013 © Yusuf Ali Yusuf 2013 I hereby declare that I am the sole author of this thesis. This is a true copy of the thesis, including any required final revisions, as accepted by my examiners. I understand that my thesis may be made electronically available to the public. ii Abstract Petroleum refining processes provide the daily requirements of energy for the global market. Each refining process produces wastes that have the capacity to harm the environment if not properly disposed of. The treatment of refinery waste is one of the most complex issues faced by refinery managers. Also, the hazardous nature of these wastes makes them rather costly to dispose of for the refineries. In this thesis, system analysis tools are used to design a program that allows for the selection of the optimal control, minimization and treating options for petroleum refinery waste streams. The performance of the developed model is demonstrated via a case study. Optimal mitigation alternatives to meet the emission reduction targets were studied by evaluating their relative impact on the profitable operation of the given facility. It was found that the optimal mitigation steps was to reduce emission precursors by conducting feed switches at the refinery. -
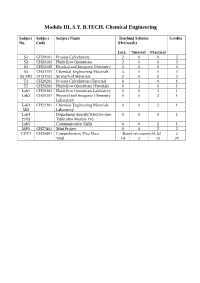
Module III, S.Y. B.TECH. Chemical Engineering
Module III, S.Y. B.TECH. Chemical Engineering Subject Subject Subject Name Teaching Scheme Credits No. Code (Hrs/week) Lect. Tutorial Practical S1 CH20101 Process Calculations 3 0 0 3 S2 CH20103 Fluid-flow Operations 3 0 0 3 S3 CH20105 Physical and Inorganic Chemistry 3 0 0 3 S4 CH21101 Chemical Engineering Materials 3 0 0 3 S5 MD CH21103 Strength of Materials 2 0 0 2 T1 CH20201 Process Calculations (Tutorial) 0 1 0 1 T2 CH20203 Fluid-flow Operations (Tutorial) 0 1 0 1 Lab1 CH20303 Fluid-flow Operations Laboratory 0 0 2 1 Lab2 CH20307 Physical and Inorganic Chemistry 0 0 2 1 Laboratory Lab3 CH21301 Chemical Engineering Materials 0 0 2 1 MD Laboratory Lab4 Department Specific Elective (see 0 0 2 1 (SD) Table after Module IV) Lab5 Communication Skills 0 0 2 1 MP3 CH27401 Mini Project 0 0 2 2 CVV1 CH20401 Comprehensive Viva Voce Based on courses S1,S2 2 Total 14 2 12 25 Issue 05: Rev No. 1: Dt. 30/03/15 FF No: 654A CH 20101:: PROCESS CALCULATIONS Credits: 03 Teaching Scheme: 3 Hours / Week Unit I: Basic Chemical Calculations (8 Hrs) A. Dimensions and Units, chemical calculations including mole, equivalent weight, solids, liquids, solutions and their properties, properties of gases. B. Significance Unit conversions of mass, energy and pressure Unit II: Material Balances Without Chemical Reactions (8 Hrs) A. Process flow sheet, Concept, Material balance calculations, recycling and bypassing operations, material balance of unsteady state processes. B. Material balance of unit operations such as distillation, crystallization Unit III: Material Balances Involving Chemical Reactions (8 Hrs) A. -

When Chemical Reactors Were Admitted and Earlier Roots of Chemical Engineering
When Chemical Reactors Were Admitted And Earlier Roots of Chemical Engineering 9 Biographical sketch of L. E. ‘Skip’ Scriven L. E. 'Skip' Scriven is Regents' Professor and holder of the L E Scriven Chair of Chemical Engineering & Materials Science at the University of Minnesota. He is a Fellow of the Minnesota Supercomputer Institute, founded the Coating Process Fundamentals Program, and now co-leads it with Professor Lorraine F. Francis. He is distinguished for pioneering researches in several areas of fluid mechanics, interfacial phenomena, porous media and surfactant technologies, and the recently emerged field of coating science and engineering. He promoted close interactions with industry by showing how good theory, incisive experimental techniques, and modern computer-aided mathematics can be combined to solve industrial processing problems. He graduated from the University of California, Berkeley, received a Ph.D. from the University of Delaware, and was a research engineer with Shell Development Company for four years before joining the University of Minnesota. He received the AIChE Allan P. Colburn Award four decades ago, the William H. Walker Award two decades ago, the Tallmadge Award in 1992, and the Founders Award in 1997. He has also been honored by the University of Minnesota and the American Society for Engineering Education for outstanding teaching. He has co-advised or advised many undergraduate, graduate and postdoctoral research students, including over 100 Ph.D.’s. Elected to the National Academy of Engineering in 1978, he has served on several U.S. national committees setting priorities for chemical engineering and materials science research. In 1990-92 he co-chaired the National Research Council's Board on Chemical Sciences and Technology, and in 1994-97 he served on the governing Commission on Physical Sciences, Mathematics, and Applications. -

Roadmap Chemical Reaction Engineering an Initiative of the Processnet Subject Division Chemical Reaction Engineering
Roadmap Chemical Reaction Engineering an initiative of the ProcessNet Subject Division Chemical Reaction Engineering 2nd edition May 2017 roadmap chemical reaction engineering Table of contents Preface 3 1 What is Chemical Reaction Engineering? 4 2 Relevance of Chemical Reaction Engineering 6 3 Experimental Reaction Engineering 8 A Laboratory Reactors 8 B High Throughput Technology 9 C Dynamic Methods 10 D Operando and in situ spectroscopic methods including spatial information 11 4 Mathematical Modeling and Simulation 14 A Challenges 14 B Workflow of Modeling and Simulation 15 C Achievements & Trends 20 5 Reactor Design and Process Development 22 A Optimization of Transport Processes in the Reactor 22 B Miniplant Technology and Experimental Scale-up 24 C Equipment Development 25 D Process Intensification 29 E Systems Engineering Approaches for Reactor Analysis, Synthesis, Operation and Control 32 6 Case studies 35 Case Study 1: The EnviNOx® Process 35 Case Study 2: Redox-Flow Batteries 36 Case Study 3: On-Board Diagnostics for Automotive Emission Control 37 Case Study 4: Simulation-based Product Design in High-Pressure Polymerization Technology 39 Case Study 5: Continuous Synthesis of Artemisinin and Artemisinin-derived Medicines 41 7 Outlook 43 Imprint 47 2 Preface This 2nd edition of the Roadmap on Chemical Reaction Engi- However, Chemical Reaction Engineering is not only driven by neering is a completely updated edition. Furthermore, it is the need for new products (market pull), but also by a rational written in English to gain more international visibility and approach to technologies (technology push). The combina- impact. Especially for the European Community of Chemical tion of digital approaches such as multiscale modeling and Reaction Engineers organized under the auspices of European simulation in combination with experimentation and space Federation of Chemical Engineering (EFCE) it can be a contri- and time-resolved in situ measurements opens up the path to bution to help to establish a Roadmap on a European level. -

PROCESS HEAT TRANSFER Course Code CH 4001 Course Title PROCESS HEAT TRANSFER Number of Credits 3 (L-2,T-1, P-0) Prerequisites NIL Course Category PC
Chemical Engineering IVSemester Prepared :2020-21 PROCESS HEAT TRANSFER Course Code CH 4001 Course Title PROCESS HEAT TRANSFER Number of Credits 3 (L-2,T-1, P-0) Prerequisites NIL Course Category PC COURSE LEARNING OBJECTIVES: • To study the fundamental concepts of heat transfer viz., conduction, convection and radiation. • To use these fundamentals in typical engineering applications (Heat exchanger and Evapora tor). COURSE OUTCOMES: On completion of the course, the student can able • to estimate steady state heat transfer rates. • to use equations for different types of convection and solve for heat transfer rate by convection •to estimate the rate of heat transfer by radiation •to estimate steam economy, capacity of single and multiple effect evaporators. COURSE CONTENTS: 1. CONDUCTION 1.1 Basic modes of heat transfer 1.2 Fourier's law of heatconduction 1.2.1 General heat conduction equations 1.2.2 Unsteady state conduction 1.2.3 Steady state conduction 1.3 Heat flow equation for composite wall 1.4 Concepts of thermal diffusivity 1.5 Heat flow equation for composite cylinder 1.6 Optimum insulation thickness 2. CONVECTION 2.1 Introduction to convection 2.2 Types of convection 2.3 Dimensional analysis 2.3.1 Criteria ofSimilitude 2.3.2 Advantages and limitations of dimensionalanalysis 2.4 Dimensionless number for heat transfer and their physicalsignificance 3. RADIATION 3.1 Thermal Radiation laws 3.2 Radiant energy distribution curve 3.3 Black and Gray bodies 3.4 Boiling 3.4.1 Boiling Phenomenon 3.4.2 Hysteresis in boiling curve 3.5 Condensation 3.5.1 Types 3.5.2 Condensation of super heated vapors 1 Chemical Engineering IVSemester Prepared :2020-21 4. -

ATTAINABLE REGION THEORY an Introduction to Choosing An
k ATTAINABLE REGION THEORY k k k k k k k k ATTAINABLE REGION THEORY An Introduction to Choosing an Optimal Reactor By DAVID MING University of the Witwatersrand, Johannesburg DAVID GLASSER and DIANE HILDEBRANDT Material and Process Synthesis research unit, University of South Africa BENJAMIN GLASSER Rutgers, The State University of New Jersey MATTHEW METZGER Merck & Co., Inc k k k k Copyright © 2016 by John Wiley & Sons, Inc. All rights reserved Published by John Wiley & Sons, Inc., Hoboken, New Jersey Published simultaneously in Canada No part of this publication may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopying, recording, scanning, or otherwise, except as permitted under Section 107 or 108 of the 1976 United States Copyright Act, without either the prior written permission of the Publisher, or authorization through payment of the appropriate per-copy fee to the Copyright Clearance Center, Inc., 222 Rosewood Drive, Danvers, MA 01923, (978) 750-8400, fax (978) 750-4470, or on the web at www.copyright.com. Requests to the Publisher for permission should be addressed to the Permissions Department, John Wiley & Sons, Inc., 111 River Street, Hoboken, NJ 07030, (201) 748-6011, fax (201) 748-6008, or online at http://www.wiley.com/go/permission. Limit of Liability/Disclaimer of Warranty: While the publisher and author have used their best efforts in preparing this book, they make no representations or warranties with respect to the accuracy or completeness of the contents of this book and specifically disclaim any implied warranties of merchantability or fitness for a particular purpose. -

Chemical Reaction Engineering A. Sarath Babu
CHEMICAL REACTION ENGINEERING A. SARATH BABU 1 Course No. Ch.E – 326 CHEMICAL REACTION ENGINEERING Periods/ Week : 4 Credits: 4 Examination Teacher Assessment: Marks: 20 Sessionals: 2 Hrs Marks: 30 End Semester: 3 Hrs Marks : 50 1. KINETICS OF HOMOGENEOUS REACTIONS 2. CONVERSION AND REACTOR SIZING 3. ANALYSIS OF RATE DATA 4. ISOTHERMAL REACTOR DESIGN 5. CATALYSIS AND CATALYTIC RECTORS 6. ADIABATIC TUBULAR REACTOR DESIGN 7. NON-IDEAL REACTORS 2 TEXT BOOKS 1. Elements of Chemical Reaction Engineering - Scott Fogler H 2. Chemical Reaction Engineering - Octave Levenspiel 3. Introduction to Chemical Reaction Engineering & Kinetics, Ronald W. Missen, Charles A. Mims, Bradley A. Saville 4. Fundamentals of Chemical Reaction Engineering – Charles D. Holland, Rayford G. Anthony 5. Chemical Reactor Analysis – R. E. Hays 6. Chemical Reactor Design and operation – K. R. Westerterp, Van Swaaij and A. A. C. M. Beenackers 7. The Engineering of Chemical Reactions – Lanny D. Schmidt 8. An Introduction to Chemical Engineering Kinetics and Reactor Design – Charles G. Hill, Jr. 9. Chemical Reactor Design, Optimization and Scaleup – E. Bruce Nauman 3 10.Reaction Kinetics and Reactor Design – John B. Butt Without chemical reaction our world would be a barren planet. No life of any sort would exist. There would be no fire for warmth and cooking, no iron and steel to make even the crudest implements, no synthetic fibers for clothing, and no engines to power our vehicles. One feature that distinguishes the chemical engineer from others is the ability to analyze systems in which chemical reactions occur and to apply the results of the analysis in a manner that benefits society. -

Process Analysis and Simulation in Chemical Engineering
Process Analysis and Simulation in Chemical Engineering Iva´n Darı´o Gil Chaves Javier Ricardo Guevara Lopez Jose´ Luis Garcı´a Zapata Alexander Leguizamon Robayo Gerardo Rodrı´guez Nino~ Process Analysis and Simulation in Chemical Engineering Iva´n Darı´o Gil Chaves Javier Ricardo Guevara Lopez Chemical and Environmental Engineering Y&V - Bohorquez Ingenierı´a Universidad Nacional de Colombia Bogota´, Colombia Bogota´, Colombia Alexander Leguizamon Robayo Jose´ Luis Garcı´a Zapata Chemical Engineering Processing Technologies Norwegian University of Science Alberta Innovates - Technology Futures and Technology Edmonton, AB, Canada Trondheim, Norway Gerardo Rodrı´guez Nino~ Chemical and Environmental Engineering National University of Colombia Bogota´, Colombia ISBN 978-3-319-14811-3 ISBN 978-3-319-14812-0 (eBook) DOI 10.1007/978-3-319-14812-0 Library of Congress Control Number: 2015955604 Springer Cham Heidelberg New York Dordrecht London © Springer International Publishing Switzerland 2016 This work is subject to copyright. All rights are reserved by the Publisher, whether the whole or part of the material is concerned, specifically the rights of translation, reprinting, reuse of illustrations, recitation, broadcasting, reproduction on microfilms or in any other physical way, and transmission or information storage and retrieval, electronic adaptation, computer software, or by similar or dissimilar methodology now known or hereafter developed. The use of general descriptive names, registered names, trademarks, service marks, etc. in this publication does not imply, even in the absence of a specific statement, that such names are exempt from the relevant protective laws and regulations and therefore free for general use. The publisher, the authors and the editors are safe to assume that the advice and information in this book are believed to be true and accurate at the date of publication.