Accurate Ab Initio Computation of Thermochemical Data for C3hx Ðx ¼ 0; ...; 4Þ Species
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

237385992.Pdf
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Radboud Repository PDF hosted at the Radboud Repository of the Radboud University Nijmegen The following full text is a publisher's version. For additional information about this publication click this link. http://hdl.handle.net/2066/208641 Please be advised that this information was generated on 2019-12-04 and may be subject to change. This is an open access article published under a Creative Commons Non-Commercial No Derivative Works (CC-BY-NC-ND) Attribution License, which permits copying and redistribution of the article, and creation of adaptations, all for non-commercial purposes. Article Cite This: J. Phys. Chem. A 2019, 123, 8053−8062 pubs.acs.org/JPCA Gas-Phase Vibrational Spectroscopy of the Hydrocarbon Cations ‑ + + ‑ + fl l C3H ,HC3H , and c C3H2 : Structures, Isomers, and the In uence of Ne-Tagging † ‡ § ∥ ‡ † ‡ ⊥ † Sandra Brünken,*, , Filippo Lipparini, , Alexander Stoffels, , Pavol Jusko, , Britta Redlich, § ‡ Jürgen Gauss, and Stephan Schlemmer † FELIX Laboratory, Institute for Molecules and Materials, Radboud University, Toernooiveld 7c, NL-6525ED Nijmegen, The Netherlands ‡ I. Physikalisches Institut, UniversitatzuKö ̈ln, Zülpicher Str. 77, D-50937 Köln, Germany § Institut für Physikalische Chemie, Johannes Gutenberg-Universitaẗ Mainz, Duesbergweg 10-14, D-55128 Mainz, Germany ∥ Dipartimento di Chimica e Chimica Industriale, Universitàdi Pisa, Via G. Moruzzi 13, I-56124 Pisa, Italy *S Supporting Information ABSTRACT: We report the first gas-phase vibrational + + spectra of the hydrocarbon ions C3H and C3H2 . The ions were produced by electron impact ionization of allene. Vibrational spectra of the mass-selected ions tagged with Ne were recorded using infrared predissociation spectroscopy in a cryogenic ion trap instrument using the intense and widely tunable radiation of a free electron laser. -

The Interstellar Chemistry of C3H and C3H2 Isomers
The interstellar chemistry of C3H and C3H2 isomers. Jean-Christophe Loison1*, Marcelino Agúndez2, Valentine Wakelam3, Evelyne Roueff4, Pierre Gratier3, Núria Marcelino5, Dianailys Nuñez Reyes1, José Cernicharo2, Maryvonne Gerin6. *Corresponding author: [email protected] 1 Institut des Sciences Moléculaires (ISM), CNRS, Univ. Bordeaux, 351 cours de la Libération, 33400, Talence, France 2 Instituto de Ciencia de Materiales de Madrid, CSIC, C\ Sor Juana Inés de la Cruz 3, 28049 Cantoblanco, Spain 3 Laboratoire d'astrophysique de Bordeaux, Univ. Bordeaux, CNRS, B18N, allée Geoffroy Saint-Hilaire, 33615 Pessac, France. 4 LERMA, Observatoire de Paris, PSL Research University, CNRS, Sorbonne Universités, UPMC Univ. Paris 06, F-92190 Meudon, France 5 INAF, Osservatorio di Radioastronomia, via P. Gobetti 101, 40129 Bologna, Italy 6 LERMA, Observatoire de Paris, PSL Research University, CNRS, Sorbonne Universités, UPMC Univ. Paris 06, Ecole Normale Supérieure, F-75005 Paris, France We report the detection of linear and cyclic isomers of C3H and C3H2 towards various starless cores and review the corresponding chemical pathways involving neutral (C3Hx with x=1,2) + and ionic (C3Hx with x = 1,2,3) isomers. We highlight the role of the branching ratio of + + electronic Dissociative Recombination (DR) reactions of C3H2 and C3H3 isomers showing * * that the statistical treatment of the relaxation of C3H and C3H2 produced in these DR reactions may explain the relative c,l-C3H and c,l-C3H2 abundances. We have also introduced in the model the third isomer of C3H2 (HCCCH). The observed cyclic-to-linear C3H2 ratio vary from 110 ± 30 for molecular clouds with a total density around 1×104 molecules.cm-3 to 30 ± 10 for molecular clouds with a total density around 4×105 molecules.cm-3, a trend well reproduced with our updated model. -

Potential Formation of Three Pyrimidine Bases in Interstellar Regions, Liton Majumdar, Prasanta Gorai, Ankan Das, Sandip
Potential formation of three pyrimidine bases in interstellar regions Liton Majumdar Univ. Bordeaux, LAB, UMR 5804, F-33270, Floirac, France CNRS, LAB, UMR 5804, F-33270, Floirac, France & Indian Centre for Space Physics, Chalantika 43, Garia Station Road, Kolkata- 700084, India Prasanta Gorai Indian Centre for Space Physics, Chalantika 43, Garia Station Road, Kolkata- 700084, India Ankan Das Indian Centre for Space Physics, Chalantika 43, Garia Station Road, Kolkata- 700084, India Sandip K. Chakrabarti S. N. Bose National Centre for Basic Sciences, Salt Lake, Kolkata- 700098, India & Indian Centre for Space Physics, Chalantika 43, Garia Station Road, Kolkata- 700084, India Received ; accepted arXiv:1511.04343v1 [physics.gen-ph] 9 Nov 2015 –2– ABSTRACT Work on the chemical evolution of pre-biotic molecules remains incomplete since the major obstacle is the lack of adequate knowledge of rate coefficients of various reactions which take place in interstellar conditions. In this work, we study the possibility of forming three pyrimidine bases, namely, cytosine, uracil and thymine in interstellar regions. Our study reveals that the synthesis of uracil from cytosine and water is quite impossible under interstellar circumstances. For the synthesis of thymine, reaction between uracil and : CH2 is investigated. Since no other relevant pathways for the formation of uracil and thymine were available in the literature, we consider a large gas-grain chemical network to study the chemical evolution of cytosine in gas and ice phases. Our modeling result shows that cytosine would be produced in cold, dense interstellar conditions. However, presence of cytosine is yet to be established. We propose that a new molecule, namely, C4N3OH5 could be observable in the interstellar region. -

I Ion Chemistry in Atmospheric and Astrophysical Plasmas
_) _// I ION CHEMISTRY IN ATMOSPHERIC AND ASTROPHYSICAL PLASMAS A. Dalgarno Harvard-Smithsonian Center for Astrophysics, 60 Garden Street, Cambridge, MA 02138, USA and J. L. Fox Institute for Terrestrial and Planetary Atmospheres, State University of New York, Stony Brook, N Y 11794, USA CONTENTS 1.1 Introduction 2 1.2 Hydrogen and Helium Plasmas 3 1.2.1 Astrophysical Environments 3 1.2.1.1 Early Universe 3 1.2.1.2 Gaseous nebulae and stellar winds 8 1.2.1.3 Supernova 1987a 9 1.2.1.4 Quasar broad-line regions 10 1.2.1.5 Dissociative shocks 10 1.2.2 Planetary Environments 12 1.2.2.1 Outer planets 12 1.3 Plasmas with an Admixture of Heavy Elements 18 1.3.1 Astrophysical Environments 18 1.3.1.1 Diffuse and translucent interstellar clouds 18 1.3.1.2 Dense molecular clouds 26 1.3.2 Outer Planets 29 Unimolecular and Bimoleeular Reaction Dynamics Edited by C. Y. Ng, T. Baer and I. Powis ,l" 1994 John Wile), & Sons Ltd A. DALGARNOAND J.L. FOX 1.4 Heavy Element Plasmas 35 1.4.1 Supernova 1987a 35 1.4.2 Terrestrial Planets 39 1.4.2.1 Dayside ionospheres 39 1.4.2.2 Nightside ionospheres 51 1.4.3 Titan and Triton 57 1.4.4 The Earth 66 1.5 Summary 76 1.1 INTRODUCTION There are many differences and also remarkable similarities between the ion chemistry and physics of planetary ionospheres and the ion chemistry and physics of astronomical environments beyond the solar system. -

Complex Organic Molecules in Solar-Type Star Forming Regions Ali Jaber Al-Edhari
Complex organic molecules in solar-type star forming regions Ali Jaber Al-Edhari To cite this version: Ali Jaber Al-Edhari. Complex organic molecules in solar-type star forming regions. Astrophysics [astro-ph]. Université Grenoble Alpes, 2016. English. NNT : 2016GREAY048. tel-01577839 HAL Id: tel-01577839 https://tel.archives-ouvertes.fr/tel-01577839 Submitted on 28 Aug 2017 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. THÈSE Pour obtenir le grade de DOCTEUR DE la Communauté UNIVERSITÉ GRENOBLE ALPES Spécialité : Astronomie & Astrophysique Arrêté ministériel : 7 Août 2006 Présentée par Ali AL-EDHARI Thèse dirigée par Cecilia Ceccarelli et codirigée par Claudine Kahane préparée au sein de l’Institut d’Astrophysique et de Planétologie de Grenoble et de l’Ecole Doctorale de Physique Complex Organic Molecules In So- lar Type Star Forming Regions Thèse soutenue publiquement le 19 Octobre 2016, devant le jury composé de : M. Eric Quirico Professeur, Université de Grenoble (France), Président Mme Paola Caselli Professeur, Max Planck Institute, MPE, Munich -

Subject Index
Cambridge University Press 978-1-107-16928-9 — Molecular Astrophysics A. G. G. M. Tielens Index More Information Subject Index ab-initio quantum chemistry, 50 anorthite, 487, 488 absorption spectroscopy, 37–41 apolar species, 33 accretion disk, 452 appearance energy, 293, 299, 302 accretion rate, 226 arenes, 33 acetylenic species, 331, 351, 402–404, 412, 512 aromatic hydrocarbons, 5, 7, 33 acid, 30, 35, 397 Aromatic Infrared Band, 258, 568–576, 592 actanoids, 23 Aromatic Infrared Band, assignments, 578 action integral, 229, 230 Aromatic Infrared Band, plateau emission, 573, 574 activity, non-ideal behavior, 142, 147 Aromatic Infrared Band, profiles, 569–572, adduct formation, 304, 312 575, 581 AGB, 402, 542, 549, 604–609 Aromatic Infrared Band, spectral fits, 575, 576 AGN toroid, 11 Aromatic Infrared Band, strength variation, AIB, see Aromatic Infrared Band 572, 573 albite, 487, 488 aromatic molecules, 397 alcohols, 7, 31, 34, 35, 397, 398 aromaticity, 260–263 aldehydes, 7, 31, 34, 35, 397, 398, 479, 508 Arrhenius law, 291 aliphatic group, 580, 584 astrobiology, 2, 4 alkali halides, 219 asymmetric top, 60, 64, 84, 118 alkali metals, 23 atomic force microscope (AFM), 46, 51 alkaline, 30 atomic number, 21, 23–25 alkaline earth metals, 23 aufbau principle, 22, 29 alkanes, 31, 32, 397, 402 autoionization, 193, 198, 269, 300 alkenes, 31, 32, 183, 261, 397, 402 alkynes, 31, 32, 183, 397, 402 B3LYP functional, 52 aluminum oxide, 145, 146, 148, 487, 488 backward reaction, 142 ambipolar diffusion, 383, 392, 394, 407, 455 bandhead, 83 ambipolar diffusion -

Burning Coal-Mining Heaps As an Organochemical Laboratory
Organic Compounds Chapter 3 Burning Coal-Mining Heaps as an Organo- chemical Laboratory: Interesting Trace Compounds and Their Potential Sources Łukasz Kruszewski* Institute of Geological Sciences, Polish Academy of Sciences, Twarda 51/55, 00-818 Warszawa, Poland. Email: [email protected] Abstract A continuation of research on the composition of complex gaseous emanations in burning post-mining waste heaps of the Upper Silesian Basin, Poland, brought new data for a large group of relatively weakly studied fumarolic vents. It concerns admixing gases either semi-quantified from in situ recorded FTIR spectra or proposed via qualitative analysis of their derivative residual curves. The most likely additional compounds include formaldoxime isocyanate, HON=CHNCO, ethenol, C2H3OH, hydrogen isocyanide, HNC, - tetrafluoro-p-xylylene, C6(CH3)2F4, 1-fluorocyclohexadienyl radical, C6H6F , perfluorinated p-benzyne, C6F4, 1,2,4-trioxolane, C2H3O3, thioacetaldehyde, CH3CHS, thiocarbonyl fluoride, dithio-p-benzoquinone, c-cyanomethanimine, bromomethane, peroxyethyl nitrate, triflic radical, CF3OSO3, and possibly a + cyclic C10 molecule. Derivatives of freons include CHClF, HCFBr, CF2I . Organo (semi)metallics include monomethylsilane, titanacyclopropene, CH3MoH and CH2MoH2, and an indium-acetylene complex. In addition, numerous inorganics may also be present. Possible pathways of the formation of the listed organics are considered. Keywords: Burning Post-Mining Waste Heaps; Coal Fires; Portable FTIR Gas Analysis; Halogenated Hydrocarbons; Gaseous Organosulfurs; Gaseous Organo (Semi) Metallics. Organic Compounds 1. Introduction Fossil fuel fires are known worldwide. They both concern completely natural environments, e.g., exposed coal or bituminous shale seams, and anthropogenic environments – burning post-coal-mining waste heaps. The latter, herein referred to as BPWHs, are, more or less, permanent elements of coal basins worldwide. -
List of Molecules and Ions with 2 to 10 Atoms
List of Molecules and Ions with 2 to 10 Atoms Diatomic (44) Molecule Designation Mass Ions AlCl Aluminium monochloride [39] [40] 62.5 — AlF Aluminium monofluoride [39] [41] 46 — AlO Aluminium monoxide [42] 43 — — Argonium [43] [44] 41 ArH+ C2 Diatomic carbon [45] [46] 24 — — Fluoromethylidynium 31 CF+[47] CH Methylidyne radical [32] [48] 13 CH+[49] CN Cyanogen radical [39] [48] [50] [51] 26 CN+,[52] CN−[53] CO Carbon monoxide [39] [54] [55] 28 CO+[56] CP Carbon monophosphide [51] 43 — CS Carbon monosulfide [39] 44 — FeO Iron(II) oxide [57] 82 — — Helium hydride ion [58] [59] 5 HeH+ H2 Molecular hydrogen [60] 2 — HCl Hydrogen chloride [61] 36.5 HCl+[62] HF Hydrogen fluoride [63] 20 — HO Hydroxyl radical [39] 17 OH+[64] KCl Potassium chloride [39] [40] 75.5 — NH Nitrogen monohydride [65] [66] 15 — N2 Molecular nitrogen [67] [68] 28 — NO Nitric oxide [69] 30 NO+[52] NS Nitrogen sulfide [39] 46 — NaCl Sodium chloride [39] [40] 58.5 — — Magnesium monohydride cation 25.3 MgH+[52] NaI Sodium iodide [70] 150 — O2 Molecular oxygen [71] 32 — PN Phosphorus mononitride [72] 45 — PO Phosphorus monoxide [73] 47 — SH Sulfur monohydride [74] 33 SH+[75] SO Sulfur monoxide [39] 48 SO+[49] SiC Carborundum [39] [76] 40 — SiN Silicon mononitride [39] 42 — SiO Silicon monoxide [39] 44 — Molecule Designation Mass Ions SiS Silicon monosulfide [39] 60 — TiO Titanium oxide [77] 63.9 — The H+ 3 cation is one of the most abundant ions in the universe. It was first detected in 1993. [78] [79] Triatomic (41) Molecule Designation Mass Ions AlNC Aluminium isocyanide -

Armando Pombeiro Coordinator CELEBRATION of the PERIODIC TABLE of the ELEMENTS at the ACADEMY of SCIENCES of LISBON. a C HEMIST
Armando Pombeiro Coordinator CELEBRATION OF THE PERIODIC TABLE OF THE ELEMENTS AT THE ACADEMY OF SCIENCES OF LISBON. A CHEMISTRY SYMPOSIUM October 3rd, 10th and 17th, 2019 FICHA TÉCNICA TÍTULO CELEBRATION OF THE PERIODIC TABLE OF THE ELEMENTS AT THE ACADEMY OF SCIENCES OF LISBON. A CHEMISTRY SYMPOSIUM COORDINATOR ARMANDO POMBEIRO EDITOR ACADEMIA DAS CIÊNCIAS DE LISBOA EDIÇÃO DIANA SARAIVA DE CARVALHO ISBN 978-972-623-394-7 ORGANIZAÇÃO Academia das Ciências de Lisboa R. Academia das Ciências, 19 1249-122 LISBOA Telefone: 213219730 Correio Electrónico: [email protected] Internet: www.acad-ciencias.pt Copyright © Academia das Ciências de Lisboa (ACL), 2020 Proibida a reprodução, no todo ou em parte, por qualquer meio, sem autorização do Editor Table of contents CELEBRATION OF THE PERIODIC TABLE OF THE ELEMENTS AT THE ACADEMY OF SCIENCES OF LISBON. A CHEMISTRY SYMPOSIUM. PREFACE. Armando J. L. Pombeiro ............................................................................................................... 1 SUBLIME GENERALIZATION: DISCOVERY OF THE PERIODIC LAW Igor S. Dmitriev and Vadim Yu. Kukushkin................................................................................. 8 CELEBRATORY SYMPOSIUM A — CATALYSIS AND THE PERIODIC TABLE HYBRID LIGANDS FOR METAL COMPLEXES, CATALYSTS AND NANOMATERIALS Pierre Braunstein ......................................................................................................................... 26 FROM A 175 YEAR OLD RUTHENIUM TO ITS EMPIRE ON GREEN CATALYSIS AND SUSTAINABLE CHEMISTRY -
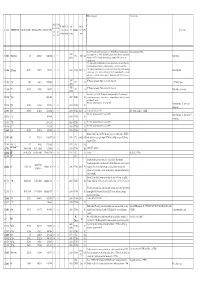
Molec Tab0410 Pub
priority for measurements ? Extra info spectro extra info astro J Max J Min K Max K Min Last lowest meas. meas. # TAG MOLECULE Min.Meas.(MHz) Max Meas.(MHz) Max Pred.(MHz) meas. meas. M. Database v level QNF Species name (or (or (or Ka) (or Ka) (entry) cm-1 N) N) The latest JPL entry includes predictions for v = 1, but otherwise appears highly similar to Detected (in list CDMS). 2007 2 13002 CH (v=0,1) 701 4250352 14289108 5 1 JPL 2733 224 that previously created in CDMS (13502) which gives predictions (for v=0 only) up to higher Methylidyne (2010) frequency (14.3 THz). Keywords: lamda-doubling, spin-rotation, LMR, infrared, not particularly rigid, * A combined fit of all available data of all isotopic species has been carried out. Note that the rotational transition measured is at different frequnency to that of previous entries. Additional rovibrational transitions between the electronic X and A states of four isotopologs 2 13503 CH+ (ion) 835137 835137 7398193 1 0 2010 CDMS 2753.6 101 Methylidynium were also used. There are two lines in the range of HIFI. Predictions should be viewed with some caution; considerable caution is advised for transitions beyond J" = 10. Keyword: ab- inito dipole moment 2 13 2008 X Π states, keywords: Hunds case(b), fine, hyperfine 2 14003 CH 7091 536132 14789069 2 1 JPL 255 13 C-Methylidyne (2009) 2008 2 2 14004 CD 439255 916954 1862237 2 1 JPL 234 X Π states, keywords: Hunds case(b), fine, hyperfine Methylidyne, deuterated (2009) New entry. -

Molecules at Z=0.89 Be Observed in Emission Because of Distance Dilution
Astronomy & Astrophysics manuscript no. paper-pks-ATCA7mm-final c ESO 2018 September 20, 2018 Molecules at z=0.89: A 4-mm-rest-frame absorption-line survey toward PKS 1830−211 S. Muller1, A. Beelen2, M. Gu´elin3,4, S. Aalto1, J. H. Black1, F. Combes5, S. J. Curran6, P. Theule7, and S. N. Longmore8 1 Onsala Space Observatory, SE 439-92, Onsala, Sweden 2 Institut d’Astrophysique Spatiale, Bˆat. 121, Universit´e Paris-Sud, 91405 Orsay Cedex, France 3 Institut de Radioastronomie Millim´etrique, 300, rue de la piscine, 38406 St Martin d’H`eres, France 4 Ecole Normale Sup´erieure/LERMA, 24 rue Lhomond, 75005 Paris, France 5 Observatoire de Paris, LERMA, CNRS, 61 Av. de l’Observatoire, 75014 Paris, France 6 School of Physics, University of New South Wales, Sydney NSW 2052, Australia 7 Physique des interactions ioniques et mol´eculaires, Universit´ede Provence, Centre de Saint J´erˆome, 13397 Marseille Cedex 20, France 8 ESO, Karl-Schwarzschild-Str. 2, 85748 Garching, Germany Received / Accepted ABSTRACT We present the results of a 7 mm spectral survey of molecular absorption lines originating in the disk of a z=0.89 spiral galaxy located in front of the quasar PKS 1830−211. Our survey was performed with the Australia Telescope Compact Array and covers the frequency interval 30–50 GHz, corresponding to the rest-frame frequency interval 57–94 GHz. A total of 28 different species, plus 8 isotopic variants, were detected toward the south-west absorption region, located about 2 kpc from the center of the z=0.89 galaxy, which therefore has the largest number of detected molecular species of any extragalactic object so far. -

The Chemistry and Spatial Distribution of Small Hydrocarbons in UV-Irradiated Molecular Clouds: the Orion Bar PDR?,?? S
A&A 575, A82 (2015) Astronomy DOI: 10.1051/0004-6361/201424568 & c ESO 2015 Astrophysics The chemistry and spatial distribution of small hydrocarbons in UV-irradiated molecular clouds: the Orion Bar PDR?;?? S. Cuadrado1;2, J. R. Goicoechea1;2, P. Pilleri3;4, J. Cernicharo1;2, A. Fuente5, and C. Joblin3;4 1 Grupo de Astrofísica Molecular, Instituto de Ciencia de Materiales de Madrid (ICMM, CSIC), Sor Juana Ines de la Cruz 3, 28049 Cantoblanco, Madrid, Spain e-mail: [email protected] 2 Centro de Astrobiología (CSIC-INTA), Carretera de Ajalvir km 4, 28850 Torrejón de Ardoz, Madrid, Spain 3 Université de Toulouse, UPS-OMP, IRAP, Toulouse, France 4 CNRS, IRAP, 9 Av. colonel Roche, BP 44346, 31028 Toulouse Cedex 4, France 5 Observatorio Astronómico Nacional, Apdo. 112, 28803 Alcalá de Henares, Madrid, Spain Received 9 July 2014 / Accepted 27 November 2014 ABSTRACT Context. Carbon chemistry plays a pivotal role in the interstellar medium (ISM) but even the synthesis of the simplest hydrocarbons and how they relate to polycyclic aromatic hydrocarbons (PAHs) and grains is not well understood. Aims. We study the spatial distribution and chemistry of small hydrocarbons in the Orion Bar photodissociation region (PDR), a prototypical environment in which to investigate molecular gas irradiated by strong UV fields. Methods. We used the IRAM 30 m telescope to carry out a millimetre line survey towards the Orion Bar edge, complemented 0 0 with ∼2 × 2 maps of the C2H and c-C3H2 emission. We analyse the excitation of the detected hydrocarbons and constrain the physi- cal conditions of the emitting regions with non-LTE radiative transfer models.