Dissolution of Iron Oxides by Oxalic Acid
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Phosphate-Based Treatments for Conservation of Stone
RILEM Technical Letters (2017) 2: 14‐19 DOI: http://dx.doi.org/10.21809/rilemtechlett.2017.34 Phosphate‐based treatments for conservation of stone Enrico Sassoni a* a Department of Civil, Chemical, Environmental and Materials Engineering, University of Bologna, Via Terracini 28, 40131, Bologna, Italy Received: 30 May 2017 / Accepted: 09 August 2017 / Published online: 9 October 2017 © The Author(s) 2017. This article is published with open access and licensed under a Creative Commons Attribution 4.0 International License. Abstract To overcome the limitations of currently available protectives and consolidants for carbonate stones (such as marble and limestone), in 2011 the use of calcium phosphate was proposed. The idea is forming calcium phosphates (ideally hydroxyapatite) as the reaction product between the substrate and an aqueous solution of a phosphate salt that the stone is treated with. In this paper, the studies aimed at identifying the best treatment conditions (in terms of nature and concentration of the phosphate precursor, solution pH, reaction time, ionic and organic additions) are first briefly summarized. Then, the efficacy of the phosphate treatment in protecting marble from dissolution in rain and restoring cohesion of weathered marble and limestone is discussed. Some recent studies on the use of the phosphate treatment on alternative substrates and some future steps for research on the topic are finally outlined. Keywords: Cultural heritage; Marble; Hydroxyapatite; Protection; Consolidation 1 Introduction improve mechanical properties, by providing a binding action between the stone grains. Organic products are A great part of cultural heritage objects (e.g. monuments, effective in improving mechanical properties, but again architectural decorations and statues) is made of carbonate they lack compatibility and durability. -

Chemistry Inventory; Fall
CHEMISTRY FALL 2005 MSDS Mfg.'s Name Chemical Name Quantity Stored Storage Conditions (on file = 9) Aluminum 9 1.5 kg Aluminum chloride, anhydrous, 98.5% 9 0.2 kg Aluminum chloride · 6H2O 9 0.5 kg Aluminum hydroxide 9 0.5 kg Aluminum nitrate 9 0.5 kg Aluminum sulfate 9 0.5 kg Ammonia, concentrated 9 4.0 L Ammonium acetate 9 0.2 kg Ammonium chloride 9 Ammonium dihydrogen phosphate (monobasic) 9 0.4 kg J.T. Baker Ammonium hydrogen phosphate (dibasic) No 0.5 kg Ammonium nitrate 9 2.5 kg Ammonium oxalate 9 0.7 kg Ammonium peroxydisulfate 9 0.5 kg Ammonium sulfate 9 0.2 kg Antimony 9 0.4 kg Barium chloride, anhydrous 9 2.5 kg Barium chloride · 2H2O 9 2.5 kg Barium nitrate 9 0.8 kg Bismuth 9 2.0 kg Boric Acid 9 0.4 kg Brass 9 Bromine 9 2.5 kg Cadmium 9 0.1 kg Cadmium nitrate 9 0.3 kg Calcium acetate · xH2O 9 0.5 kg Calcium carbide 9 1.0 kg Calcium carbonate 9 2.2 kg Calcium chloride 9 1.0 kg Calcium hydroxide 9 0.3 kg Calcium nitrate · 4H2O 9 1.0 kg Calcium oxide 9 0.3 kg Calcium sulfate · 2H2O 9 1.0 kg Carbon 9 0.1 kg Ceric ammonium nitrate 9 0.5 kg Cesium chloride 9 0.01 kg Chromium 9 0.01 kg Chromium chloride 9 0.5 kg Chromium nitrate 9 0.5 kg Cobalt 9 0.025 kg Cobalt chloride 9 0.7 kg Cobalt nitrate 9 0.6 kg Copper (assorted) 9 4.0 kg Copper acetate 9 0.05 kg Copper chloride 9 0.1 kg Copper nitrate 9 3.5 kg Copper oxide 9 0.4 kg Cupric sulfate, anhydrous 9 0.5 kg Cupric sulfate · 5H2O 9 2.75 kg EDTA 9 0.6 kg Iodine 9 2.0 kg Iron (assorted) 9 5.0 kg MSDS Mfg.'s Name Chemical Name Quantity Stored Storage Conditions (on file = 9) Ferric ammonium -
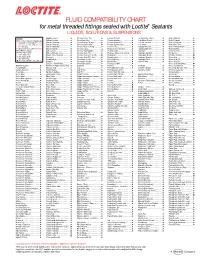
FLUID COMPATIBILITY CHART for Metal Threaded Fittings Sealed with Loctite¨ Sealants LIQUIDS, SOLUTIONS & SUSPENSIONS
FLUID COMPATIBILITY CHART for metal threaded fittings sealed with Loctite® Sealants LIQUIDS, SOLUTIONS & SUSPENSIONS LEGEND: Bagasse Fibers.......................... Chlorobenzene Dry ................... Ferrous Chloride ...................... Ion Exclusion Glycol ................. Nickel Chloride.......................... All Loctite® Anaerobic Sealants are Barium Acetate ........................ Chloroform Dry......................... Ferrous Oxalate......................... Irish Moss Slurry...................... Nickel Cyanide ......................... Compatible Including #242®, 243, Barium Carbonate..................... Chloroformate Methyl............... Ferrous Sulfate10%.................. Iron Ore Taconite ..................... Nickel Fluoborate ..................... 542, 545, 565, 567, 569, 571, 572, Barium Chloride........................ Chlorosulfonic Acid .................. Ferrous Sulfate (Sat)................. Iron Oxide ................................ Nickel Ore Fines ....................... 577, 580, 592 Barium Hydroxide..................... Chrome Acid Cleaning .............. Fertilizer Sol ............................. Isobutyl Alcohol ....................... Nickel Plating Bright ................. † Use Loctite® #270, 271™, 277, 554 Barium Sulfate.......................... Chrome Liquor.......................... Flotation Concentrates.............. Isobutyraldehyde ..................... Nickel Sulfate ........................... Not Recommended Battery Acid .............................. Chrome Plating -

Ammonium Phosphate on Gypsum
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by AMS Acta - Alm@DL - Università di Bologna HMC 2016 4 th Historic Mortars Conference Preliminary study on the use of ammonium phosphate for the conservation of marble-imitating gypsum-stuccoes Enrico Sassoni 1, Gabriela Graziani 2, George W. Scherer 3 and Elisa -ranzoni 4 0Tmh dqrhsxneAnknfm+)skx+ [email protected] 1Tmh dqrhsxneAnknfm+)skx+ faqhdk-fqyhmh1.tmhan-hs 2 OqhmbdsnmTmh dqrhsx+MI+TR@+ rbgdqdq.oqhmbdsnm-dct 0 Tmh dqrhsxneAnknfm+)skx+ dkhr-eqmynmh.tmhan-hs Abstract6 In this study, a novel method for consolidation and im rovement of resistance to water of gy sum-stuccoes was reliminarily investigated. The idea is treating gy sum with an aqueous solution of diammonium hydrogen hos hate (DAP, (.H 4)2HPO 4) to form hydroxya atite (HAP, Ca 10 (PO 4)6(OH) 2), which has much lower solubility than gy sum. Tests carried out on gy sum aste sam les, manufactured to resemble historic stuccoes, showed that, after treatment with the DAP solution, a significant im rovement in mechanical ro erties was achieved and brushite (CaHPO 4ì2H 2O) was formed (alongside some other by- roducts, that can be removed by an additional oultice treatment). Even if brushite is more soluble than HAP, still its formation is ex ected to be beneficial for stuccoes conservation, as brushite is significantly less soluble than gy sum. Introduction Since antiquity, gy sum-based stuccoes have been frequently used to imitate recious white or colored marbles, when the use of real marbles was not ossible, because trans ort from faraway quarries was too costly a1,2b. -

Dictionary of Explosives
DICTIONARY OF EXPLOSIVES BY ARTHUR MARSHALL A .C .G j., F.I.C., F.C.S. CHEMICAL INSPECTOR INDIAN ORDNANCE DEPARTMENT PHILADELPHIA P. BLAKISTON’S SON & CO. 1012 WALNUT STREET 1920 Printed in Great Britain INTRODUCTION It is a generation since a dictionary of explosives has been published, and, in the meantime, many new explosives have been introduced. It is hoped, therefore, that this small volume, giving concise information about these special materials, may prove useful to those who have to deal with them. In Cundill and Thomson’s “ Dictionary of Explosives,” issued in 1895, there arc many entries of the names of inventors and of mixtures which had been proposed but have never been used commercially, nor are likely to be. As modem explosives were then in their infancy, it was no doubt wise to insert all the available information whether it appeared to be important or not; but now it seems to me better to restrict the scope of the dictionary so as to keep its size within moderate limits. Practically only explosives with special or proprietary names are therefore dealt with here. For information concerning chemical substances, such as the nitro-toluenes and other nitro-compounds, reference should be made to the text-books on explosives and chemistry. A few words may, however, be said here about the nitro- celluloses. These are made by treating cellulose with a mix ture of nitric and sulphuric acids, and then purifying the product by washing it thoroughly with hot water. The variety of cellulose most used for this purpose is cotton, and the product obtained from it is frequently called nitrocotton, three special varieties of which are collodion cotton, pyro- collodion and guncotton (q. -

Safety Data Sheet
SAFETY DATA SHEET Preparation Date: 1/12/2016 Revision Date: 2/13/2018 Revision Number: G2 1. IDENTIFICATION Product identifier Product code: F1002 Product Name: FERRIC AMMONIUM OXALATE, GRANULAR Other means of identification Synonyms: Ethanedioic acid, ammonium iron(3+) salt (3:3:1), trihydrate Ethanedioic acid, iron (3+) ammonium salt, hydrate (3:1:3:3) Oxalic acid, ammonium iron (3+) salt (3:3:1), trihydrate Iron ammonium oxalate, (NH4)3Fe(C2O4)3, trihydrate Ammonium iron oxalate, (NH4)3Fe(C2O4)3, trihydrate CAS #: 13268-42-3 RTECS # LJ8932000 (for anhydrous form CAS no. 14221-47-7) CI#: Not available Recommended use of the chemical and restrictions on use Recommended use: In photography. blueprpints. Coloring of aluminum and aluminum alloys. Uses advised against No information available Supplier: Spectrum Chemical Mfg. Corp 14422 South San Pedro St. Gardena, CA 90248 (310) 516-8000 Order Online At: https://www.spectrumchemical.com Emergency telephone number Chemtrec 1-800-424-9300 Contact Person: Martin LaBenz (West Coast) Contact Person: Ibad Tirmiz (East Coast) 2. HAZARDS IDENTIFICATION Classification This chemical is considered hazardous according to the 2012 OSHA Hazard Communication Standard (29 CFR 1910.1200) Considered a dangerous substance or mixture according to the Globally Harmonized System (GHS) Acute toxicity - Oral Category 4 Acute toxicity - Dermal Category 4 Skin corrosion/irritation Category 2 Serious eye damage/eye irritation Category 2 Specific target organ toxicity (single exposure) Category 3 Label elements Warning -

Determination of Titanium, Zirconium, Niobium, and Tantalum in Steels: Separations by Anion-Exchange John 1
Journal of Research of the National Bureau of Standards Vo!' 62, No.1, January 1959 Research Paper 2923 Determination of Titanium, Zirconium, Niobium, and Tantalum in Steels: Separations by Anion-Exchange John 1. Hague and Lawrence A . Machlan A procedlll'e is described for the determination of titanium, zirconium niobium and tantalum in steel. These elements are concentrated by selective precipita'tion with' cup ferr on from a hydrochloric acid solution of the sample, and t hen ignited to t he oxides. The oXides are fused, and dissolved in a hydrochloric-hydrofluoric acid m ixtlll'e. Three sepa rate eluates, containing titanium and zirconium, niobium, and tantalum result from elu tion with mixtures containing ammonium chloride, hydrochloric acid, and hydrofluoric acid from a column of Dowex-l anion-exchange resin. Boric acid is added to the clu ates to complex t he hydrofluoric acid, and the elements are precipitated wit h cupferron. Titanium is deter mmed by the hydrogen-peroxide method, and zirconium by the phosphate-gravimetric method. NIOblllm and tantalum are determl11ed by the hydroquinone- and pyrogallol photometflC methods, or by welghl11g the oX ides. 1. Introduction A concentration of Litanium, zil'co llinum, Iliobiulll, and tantalum is eff ected by cupfeJ'l'oLl prcci pitation T he incrcase d usC' in recent years of clements [4, 5] with most of the iron present in the reduced t hat form complexes with carbo;, and nitroO'en in co ndition. The small amount of iron coprecipit ated metallurgical alloys has presented some inte;esti ll g serves as a gathering agent. -

Effects of Impurities on an Industrial Crystallization Process of Ammonium Sulfate
Effects of impurities on an industrial crystallization process of ammonium sulfate Dissertation zur Erlangung des akademischen Grades Doktoringenieur (Dr.-Ing.) vorgelegt dem Zentrum für Ingenieurwissenschaften der Martin-Luther-Universität Halle-Wittenberg als organisatorische Grundeinheit für Forschung und Lehre im Range einer Fakultät (§ 75 Abs. 1 HSG LSA, § 1 Abs. 1 Grundordnung) von Herrn Dipl.-Ing. Robert Buchfink geboren am 18.04.1983 Gutachter: 1. Prof. Dr.-Ing. Dr. h.c. Joachim Ulrich 2. Prof. Dr. rer. nat. habil. Axel König Datum der Verteidigung: 02.05.2011 Halle (Saale), den 01.06.2011 Acknowledgment First of all I want to thank my family including my parents Gunter und Sigrid, my sister Lydia and my grandfather Walter. I wished my grandmother Hanni could have seen me getting a PhD degree. All of them gave me a great support, both financial and moral, over the long period of study and up to now we have a fantastic relationship. Furthermore, I want to acknowledge the support of my girlfriend Elli who was always there for me in good and in bad times. No PhD thesis without a supervisor, an institute and a topic. In this context I want to thank Prof. Dr.-Ing. Dr. h.c. Joachim Ulrich who gave me the great opportunity to work at his institute and who supervised my research in the interesting field of crystallization. Moreover, I also want to thank Prof. Ulrich for creating the institute as a melting pot of international students which opened my mind in many ways and gave me the opportunity to get close friendships and learn much about the culture of different nations. -

List of Lists
United States Office of Solid Waste EPA 550-B-10-001 Environmental Protection and Emergency Response May 2010 Agency www.epa.gov/emergencies LIST OF LISTS Consolidated List of Chemicals Subject to the Emergency Planning and Community Right- To-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act • EPCRA Section 302 Extremely Hazardous Substances • CERCLA Hazardous Substances • EPCRA Section 313 Toxic Chemicals • CAA 112(r) Regulated Chemicals For Accidental Release Prevention Office of Emergency Management This page intentionally left blank. TABLE OF CONTENTS Page Introduction................................................................................................................................................ i List of Lists – Conslidated List of Chemicals (by CAS #) Subject to the Emergency Planning and Community Right-to-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act ................................................. 1 Appendix A: Alphabetical Listing of Consolidated List ..................................................................... A-1 Appendix B: Radionuclides Listed Under CERCLA .......................................................................... B-1 Appendix C: RCRA Waste Streams and Unlisted Hazardous Wastes................................................ C-1 This page intentionally left blank. LIST OF LISTS Consolidated List of Chemicals -

Technical Method
J Clin Pathol: first published as 10.1136/jcp.29.11.1038 on 1 November 1976. Downloaded from 1038 Technical method Technical method Simplified qualitative chemical the phosphate test: first add two drops of a 10%° ammonium hydroxide solution, one drop of 20 % analysis for urinary calculi nitric acid, and then two to three drops of the ammonium-molybdate reagent. Heating the J. KLEEBERG mixture will produce the characteristic yellow Research Department, Rothschild- UniversityHospital, colour ofammonium-molybdate-phosphate. Haifa, Israel The 'three glass slide' technique to be described here SLIDE II TEST FOR OXALATE is meant for qualitative stone analysis only and Two to three milligrams of stone powderismixedwith applies especially to small calculi. double the amount of resorcinol powder and 2 drops of concentrated sulphuric acid, and the mixture is Material and methods heated. The appearance of a deep blue (not green!) colour is proof of oxalate (Dorronsorro and Bi-distilled water; acetic acid 10%; hydrochloric Fernandez, cited by Higgins (1954)). If oxalate is acid 10%; nitric acid 20%; sulphuric acid conc; found, calcium must also be present. (Resorcinol and ammonium hydroxide solution 10%; sodium sulphuric acid produce coloured compounds with hydroxide solution 10 %; resorcinol powder; satura- dicarbonic acids (Feigl, 1956).) ted aqueous solution of ammonium-molybdate. Magneson reagent: 1 mg of nitro-phenyl-azo- SLIDE III TESTS FOR AMMONIA, CARBONATE, resorcinol is dissolved in 100 ml of 2-N-sodium MAGNESIUM AND CALCIUM hydroxide. (a) 2-3 mg of stone powder is moistened with 2 dropscopyright. of 10% hydrochloric acid, gently warmed, and SLIDE I TESTS FOR AMMONIUM URATE, URIC examined for bubbles with a magnifying glass: ACID, CYSTINE, XANTHINE, AND PHOSPHATE evidence ofcarbonate. -
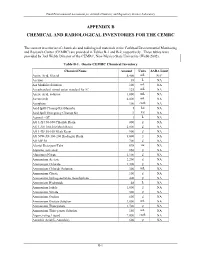
Appendix B Chemical and Radiological Inventories for the Cemrc
Final Environmental Assessment for Actinide Chemistry and Repository Science Laboratory APPENDIX B CHEMICAL AND RADIOLOGICAL INVENTORIES FOR THE CEMRC The current inventories of chemicals and radiological materials at the Carlsbad Environmental Monitoring and Research Center (CEMRC) are provided in Tables B-1 and B-2, respectively. These tables were provided by Joel Webb, Director of the CEMRC, New Mexico State University (Webb 2002). Table B-1. Onsite CEMRC Chemical Inventory Chemical Name Amount Units SARA Limit Acetic Acid, Glacial 5,400 mL NAa Acetone 38 L NA AA Modifier Solution 100 mL NA AccuStandard mixed anion standard for IC 125 mL NA Acetic Acid, solution 1,000 mL NA Acetonitrile 4,000 mL NA Acetylene 100 cu.ft. NA Acid Spill Cleanup Kit (Hazorb) 1 kit NA Acid Spill Emergency Cleanup Kit 3 kit NA Aerosol - OT 1 L NA AG 1-X4 50-100 Chloride Resin 800 g NA AG 1-X8 100-200 Mesh Resin 2,000 g NA AG 1-X8 50-100 Mesh Resin 900 g NA AG 50W-X8 100-200 Hydrogen Resin 3,000 g NA AG MP 50 700 g NA Alcojet Detergent/Tabs 698 oz NA Alumina, activated 850 g NA Aluminum Nitrate 2,100 g NA Ammonium Acetate 2,200 g NA Ammonium Chloride 1,300 g NA Ammonium Chloride Solution 300 mL NA Ammonium Citrate 100 g NA Ammonium hydrogenoxalate, hemihydrate 400 g NA Ammonium Hydroxide 48 L NA Ammonium Iodide 1,000 g NA Ammonium Nitrate 500 g NA Ammonium Oxalate 600 g NA Ammonium Oxalate Solution 2,000 mL NA Ammonium Thiocyanate 1,700 g NA Ammonium Thiocyanate Solution 150 mL NA Argon, refrig. -

Chemistry of Strontium in Natural Water
Chemistry of Strontium in Natural Water GEOLOGICAL SURVEY WATER-SUPPLY PAPER 1496 This water-supply paper was printed as separate chapters A-D UNITED STATES GOVERNMENT PRINTING OFFICE, WASHINGTON : 1963 UNITED STATES DEPARTMENT OF THE INTERIOR STEWART L. UDALL, Secretary GEOLOGICAL SURVEY Thomas B. Nolan, Director The U.S. Geological Survey Library has cataloged this publication as follows: U.S. Geological Survey. Chemistry of strontium in natural water. Washington, U.S. Govt. Print. Off., 1962. iii, 97 p. illus., diagrs., tables. 24 cm. (Its Water-supply paper 1496) Issued as separate chapters A-D. Includes bibliographies. 1. Strontium. 2. Water-Analysis. I. Title. (Series) CONTENTS [The letters in parentheses preceding the titles are those used to designate the separate chapters] Page (A) A survey of analytical methods for the determination of strontium in natural water, by C. Albert Horr____________________________ 1 (B) Copper-spark method for spectrochemical determination of strontirm in water, by Marvin W. Skougstad-______-_-_-_--_~__-___-_- 19 (C) Flame photometric determination of strontium in natural water, by C. Albert Horr_____._____._______________... 33 (D) Occurrence and distribution of strontium in natural water, by Margin W. Skougstad and C. Albert Horr____________.___-._-___-. 55 iii A Survey of Analytical Methods fc r The Determination of Strontium in Natural Water By C. ALBERT HORR CHEMISTRY OF STRONTIUM IN NATURAL rVATER GEOLOGICAL SURVEY WATER-SUPPLY PAPER 1496-A This report concerns work done on behalf of the U.S. Atomic Energy Commission and is published with the permission of the Commission UNITED STATES GOVERNMENT PRINTING OFFICE, WASHINGTON : 1959 UNITED STATES DEPARTMENT OF THE INTERIOR FRED A.