Download/Tandemms.Doc)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Capacity of Long-Term in Vitro Proliferation of Acute Myeloid
The Capacity of Long-Term in Vitro Proliferation of Acute Myeloid Leukemia Cells Supported Only by Exogenous Cytokines Is Associated with a Patient Subset with Adverse Outcome Annette K. Brenner, Elise Aasebø, Maria Hernandez-Valladares, Frode Selheim, Frode Berven, Ida-Sofie Grønningsæter, Sushma Bartaula-Brevik and Øystein Bruserud Supplementary Material S2 of S31 Table S1. Detailed information about the 68 AML patients included in the study. # of blasts Viability Proliferation Cytokine Viable cells Change in ID Gender Age Etiology FAB Cytogenetics Mutations CD34 Colonies (109/L) (%) 48 h (cpm) secretion (106) 5 weeks phenotype 1 M 42 de novo 241 M2 normal Flt3 pos 31.0 3848 low 0.24 7 yes 2 M 82 MF 12.4 M2 t(9;22) wt pos 81.6 74,686 low 1.43 969 yes 3 F 49 CML/relapse 149 M2 complex n.d. pos 26.2 3472 low 0.08 n.d. no 4 M 33 de novo 62.0 M2 normal wt pos 67.5 6206 low 0.08 6.5 no 5 M 71 relapse 91.0 M4 normal NPM1 pos 63.5 21,331 low 0.17 n.d. yes 6 M 83 de novo 109 M1 n.d. wt pos 19.1 8764 low 1.65 693 no 7 F 77 MDS 26.4 M1 normal wt pos 89.4 53,799 high 3.43 2746 no 8 M 46 de novo 26.9 M1 normal NPM1 n.d. n.d. 3472 low 1.56 n.d. no 9 M 68 MF 50.8 M4 normal D835 pos 69.4 1640 low 0.08 n.d. -

Chuanxiong Rhizoma Compound on HIF-VEGF Pathway and Cerebral Ischemia-Reperfusion Injury’S Biological Network Based on Systematic Pharmacology
ORIGINAL RESEARCH published: 25 June 2021 doi: 10.3389/fphar.2021.601846 Exploring the Regulatory Mechanism of Hedysarum Multijugum Maxim.-Chuanxiong Rhizoma Compound on HIF-VEGF Pathway and Cerebral Ischemia-Reperfusion Injury’s Biological Network Based on Systematic Pharmacology Kailin Yang 1†, Liuting Zeng 1†, Anqi Ge 2†, Yi Chen 1†, Shanshan Wang 1†, Xiaofei Zhu 1,3† and Jinwen Ge 1,4* Edited by: 1 Takashi Sato, Key Laboratory of Hunan Province for Integrated Traditional Chinese and Western Medicine on Prevention and Treatment of 2 Tokyo University of Pharmacy and Life Cardio-Cerebral Diseases, Hunan University of Chinese Medicine, Changsha, China, Galactophore Department, The First 3 Sciences, Japan Hospital of Hunan University of Chinese Medicine, Changsha, China, School of Graduate, Central South University, Changsha, China, 4Shaoyang University, Shaoyang, China Reviewed by: Hui Zhao, Capital Medical University, China Background: Clinical research found that Hedysarum Multijugum Maxim.-Chuanxiong Maria Luisa Del Moral, fi University of Jaén, Spain Rhizoma Compound (HCC) has de nite curative effect on cerebral ischemic diseases, *Correspondence: such as ischemic stroke and cerebral ischemia-reperfusion injury (CIR). However, its Jinwen Ge mechanism for treating cerebral ischemia is still not fully explained. [email protected] †These authors share first authorship Methods: The traditional Chinese medicine related database were utilized to obtain the components of HCC. The Pharmmapper were used to predict HCC’s potential targets. Specialty section: The CIR genes were obtained from Genecards and OMIM and the protein-protein This article was submitted to interaction (PPI) data of HCC’s targets and IS genes were obtained from String Ethnopharmacology, a section of the journal database. -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -

Identification of Novel Alleles Associated with Insulin Resistance In
OPEN International Journal of Obesity (2018) 42, 686–695 www.nature.com/ijo PEDIATRIC ORIGINAL ARTICLE Identification of novel alleles associated with insulin resistance in childhood obesity using pooled-DNA genome-wide association study approach P Kotnik1,7, E Knapič1,2,7, J Kokošar3, J Kovač4, R Jerala5, T Battelino1,6 and S Horvat2,5 BACKGROUND: Recently, we witnessed great progress in the discovery of genetic variants associated with obesity and type 2 diabetes (T2D), especially in adults. Much less is known regarding genetic variants associated with insulin resistance (IR). We hypothesized that novel IR genes could be efficiently detected in a population of obese children and adolescents who may not exhibit comorbidities and other confounding factors. OBJECTIVES: This study aimed to determine whether a genome-wide association study (GWAS), using a DNA-pooling approach, could identify novel genes associated with IR. SUBJECTS: The pooled-DNA GWAS analysis included Slovenian obese children and adolescents with and without IR matched for body mass index, gender and age. A replication study was conducted in another independent cohort with or without IR. METHODS: For the pooled-DNA GWAS, we used HumanOmni5-Quad SNP array (Illumina). Allele frequency distributions were compared with modified t-tests and χ2-tests and ranked using PLINK. Top single nucleotide polymorphisms (SNPs) were validated using individual genotyping by high-resolution melting analysis and TaqMan assay. RESULTS: We identified five top-ranking SNPs from the pooled-DNA GWAS analysis within the ECE1, IL1R2, GNPDA1, HLA-J and PYGB loci. All except SNP rs9261108 (HLA-J locus) were confirmed in the validation phase using individual genotyping. -

A Novel PLEKHA7 Interactor at Adherens Junctions
Thesis PDZD11: a novel PLEKHA7 interactor at adherens junctions GUERRERA, Diego Abstract PLEKHA7 is a recently identified protein of the AJ that has been involved by genetic and genomic studies in the regulation of miRNA signaling and cardiac contractility, hypertension and glaucoma. However, the molecular mechanisms behind PLEKHA7 involvement in tissue physiology and pathology remain unknown. In my thesis I report novel results which uncover PLEKHA7 functions in epithelial and endothelial cells, through the identification of a novel molecular interactor of PLEKHA7, PDZD11, by yeast two-hybrid screening, mass spectrometry, co-immunoprecipitation and pulldown assays. I dissected the structural basis of their interaction, showing that the WW domain of PLEKHA7 binds to the N-terminal region of PDZD11; this interaction mediates the junctional recruitment of PDZD11, identifying PDZD11 as a novel AJ protein. I provided evidence that PDZD11 forms a complex with nectins at AJ, its PDZ domain binds to the PDZ-binding motif of nectins. PDZD11 stabilizes nectins promoting the early steps of junction assembly. Reference GUERRERA, Diego. PDZD11: a novel PLEKHA7 interactor at adherens junctions. Thèse de doctorat : Univ. Genève, 2016, no. Sc. 4962 URN : urn:nbn:ch:unige-877543 DOI : 10.13097/archive-ouverte/unige:87754 Available at: http://archive-ouverte.unige.ch/unige:87754 Disclaimer: layout of this document may differ from the published version. 1 / 1 UNIVERSITE DE GENÈVE FACULTE DES SCIENCES Section de Biologie Prof. Sandra Citi Département de Biologie Cellulaire PDZD11: a novel PLEKHA7 interactor at adherens junctions THÈSE Présentée à la Faculté des sciences de l’Université de Genève Pour obtenir le grade de Doctor ès science, mention Biologie par DIEGO GUERRERA de Benevento (Italie) Thèse N° 4962 GENÈVE Atelier d'impression Repromail 2016 1 Table of contents RÉSUMÉ .................................................................................................................. -

Exploring the Mechanism of Shengmai Yin for Coronary Heart Disease Based on Systematic Pharmacology and Chemoinformatics
Bioscience Reports (2020) 40 BSR20200286 https://doi.org/10.1042/BSR20200286 Research Article Exploring the mechanism of Shengmai Yin for coronary heart disease based on systematic pharmacology and chemoinformatics Yan Jiang1,2,*,QiHe3,*, Tianqing Zhang4,5,*,WangXiang6,7, Zhiyong Long8,9 and Shiwei Wu10 1Department of General Surgery, The First Affiliated Hospital of University of South China, Hengyang, Hunan Province, China; 2Graduate College, Hunan Normal University, Changsha, Hunan Province, China; 3Intensive Care Unit, People’s Hospital of Ningxiang City, Ningxiang 410600, Hunan Province, China; 4 Graduate College, University of South China, Hengyang, Hunan Province, China; 5Department of Cardiology, The First Affiliated Hospital of University of South China, Hengyang, Hunan Province, China; 6Graduate College, Guilin Medical University, Guilin, Guangxi Province, China; 7Department of Rheumatology, Affiliated Hospital of Guilin Medical University, Guilin, Guangxi Province, China; 8Department of Physical Medicine and Rehabilitation, Guangdong General Hospital, Shantou University Medical College, Shantou, Guangdong, China; 9Graduate College, Shantou University Medical College, Shantou, Guangdong Province, China; 10Department of Traditional Chinese Medicine, The Eighth Affiliated Hospital, Sun Yat-sen University, Shenzhen, Guangdong Province, China Correspondence: Shiwei Wu ([email protected]) Objective: To explore the mechanism of Shengmai Yin (SMY) for coronary heart disease (CHD) by systemic pharmacology and chemoinformatics. Methods: Traditional Chinese Medicine Systems Pharmacology Database (TCMSP), tradi- tional Chinese medicine integrative database (TCMID) and the traditional Chinese medicine (TCM) Database@Taiwan were used to screen and predict the bioactive components of SMY. Pharmmapper were utilized to predict the potential targets of SMY, the TCMSP was utilized to obtain the known targets of SMY. The Genecards and OMIM database were utilized to collect CHD genes. -
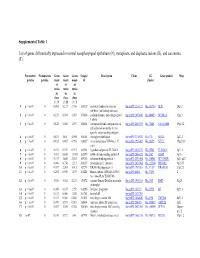
Supplemental Table 1 List of Genes Differentially Expressed In
Supplemental Table 1 List of genes differentially expressed in normal nasopharyngeal epithelium (N), metaplastic and displastic lesions (R), and carcinoma (T). Parametric Permutation Geom Geom Geom Unique Description Clone UG Gene symbol Map p-value p-value mean mean mean id cluster of of of ratios ratios ratios in in in class class class 1 : N 2 : R 3 : T 1 p < 1e-07 0 0.061 0.123 2.708 169329 secretory leukocyte protease IncytePD:2510171 Hs.251754 SLPI 20q12 inhibitor (antileukoproteinase) 2 p < 1e-07 0 0.125 0.394 1.863 163628 sodium channel, nonvoltage-gated IncytePD:1453049 Hs.446415 SCNN1A 12p13 1 alpha 3 p < 1e-07 0 0.122 0.046 1.497 160401 carcinoembryonic antigen-related IncytePD:2060355 Hs.73848 CEACAM6 19q13.2 cell adhesion molecule 6 (non- specific cross reacting antigen) 4 p < 1e-07 0 0.675 1.64 5.594 165101 monoglyceride lipase IncytePD:2174920 Hs.6721 MGLL 3q21.3 5 p < 1e-07 0 0.182 0.487 0.998 166827 nei endonuclease VIII-like 1 (E. IncytePD:1926409 Hs.28355 NEIL1 15q22.33 coli) 6 p < 1e-07 0 0.194 0.339 0.915 162931 hypothetical protein FLJ22418 IncytePD:2816379 Hs.36563 FLJ22418 1p11.1 7 p < 1e-07 0 1.313 0.645 13.593 162399 S100 calcium binding protein P IncytePD:2060823 Hs.2962 S100P 4p16 8 p < 1e-07 0 0.157 1.445 2.563 169315 selenium binding protein 1 IncytePD:2591494 Hs.334841 SELENBP1 1q21-q22 9 p < 1e-07 0 0.046 0.738 1.213 160115 prominin-like 1 (mouse) IncytePD:2070568 Hs.112360 PROML1 4p15.33 10 p < 1e-07 0 0.787 2.264 3.013 167294 HRAS-like suppressor 3 IncytePD:1969263 Hs.37189 HRASLS3 11q12.3 11 p < 1e-07 0 0.292 0.539 1.493 168221 Homo sapiens cDNA FLJ13510 IncytePD:64451 Hs.37896 2 fis, clone PLACE1005146. -

Co-Evolution of B7H6 and Nkp30, Identification of a New B7 Family Member, B7H7, and of B7's Historical Relationship with the MHC
Immunogenetics (2012) 64:571–590 DOI 10.1007/s00251-012-0616-2 ORIGINAL PAPER Evolution of the B7 family: co-evolution of B7H6 and NKp30, identification of a new B7 family member, B7H7, and of B7's historical relationship with the MHC Martin F. Flajnik & Tereza Tlapakova & Michael F. Criscitiello & Vladimir Krylov & Yuko Ohta Received: 19 January 2012 /Accepted: 20 March 2012 /Published online: 11 April 2012 # Springer-Verlag 2012 Abstract The B7 family of genes is essential in the regula- Furthermore, we identified a Xenopus-specific B7 homolog tion of the adaptive immune system. Most B7 family mem- (B7HXen) and revealed its close linkage to B2M, which we bers contain both variable (V)- and constant (C)-type have demonstrated previously to have been originally domains of the immunoglobulin superfamily (IgSF). encoded in the MHC. Thus, our study provides further proof Through in silico screening of the Xenopus genome and that the B7 precursor was included in the proto MHC. subsequent phylogenetic analysis, we found novel genes Additionally, the comparative analysis revealed a new B7 belonging to the B7 family, one of which is the recently family member, B7H7, which was previously designated in discovered B7H6. Humans and rats have a single B7H6 the literature as an unknown gene, HHLA2. gene; however, many B7H6 genes were detected in a single large cluster in the Xenopus genome. The B7H6 expression Keywords B7 family. MHC . Evolution . Natural killer patterns also varied in a species-specific manner. Human receptors . Genetic linkage . Xenopus B7H6 binds to the activating natural killer receptor, NKp30. While the NKp30 gene is single-copy and maps to the MHC in most vertebrates, many Xenopus NKp30 genes were Introduction found in a cluster on a separate chromosome that does not harbor the MHC. -

The Human Gene Connectome As a Map of Short Cuts for Morbid Allele Discovery
The human gene connectome as a map of short cuts for morbid allele discovery Yuval Itana,1, Shen-Ying Zhanga,b, Guillaume Vogta,b, Avinash Abhyankara, Melina Hermana, Patrick Nitschkec, Dror Friedd, Lluis Quintana-Murcie, Laurent Abela,b, and Jean-Laurent Casanovaa,b,f aSt. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY 10065; bLaboratory of Human Genetics of Infectious Diseases, Necker Branch, Paris Descartes University, Institut National de la Santé et de la Recherche Médicale U980, Necker Medical School, 75015 Paris, France; cPlateforme Bioinformatique, Université Paris Descartes, 75116 Paris, France; dDepartment of Computer Science, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel; eUnit of Human Evolutionary Genetics, Centre National de la Recherche Scientifique, Unité de Recherche Associée 3012, Institut Pasteur, F-75015 Paris, France; and fPediatric Immunology-Hematology Unit, Necker Hospital for Sick Children, 75015 Paris, France Edited* by Bruce Beutler, University of Texas Southwestern Medical Center, Dallas, TX, and approved February 15, 2013 (received for review October 19, 2012) High-throughput genomic data reveal thousands of gene variants to detect a single mutated gene, with the other polymorphic genes per patient, and it is often difficult to determine which of these being of less interest. This goes some way to explaining why, variants underlies disease in a given individual. However, at the despite the abundance of NGS data, the discovery of disease- population level, there may be some degree of phenotypic homo- causing alleles from such data remains somewhat limited. geneity, with alterations of specific physiological pathways under- We developed the human gene connectome (HGC) to over- come this problem. -

Meta-Analysis of Gene Expression in Individuals with Autism Spectrum Disorders
Meta-analysis of Gene Expression in Individuals with Autism Spectrum Disorders by Carolyn Lin Wei Ch’ng BSc., University of Michigan Ann Arbor, 2011 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF Master of Science in THE FACULTY OF GRADUATE AND POSTDOCTORAL STUDIES (Bioinformatics) The University of British Columbia (Vancouver) August 2013 c Carolyn Lin Wei Ch’ng, 2013 Abstract Autism spectrum disorders (ASD) are clinically heterogeneous and biologically complex. State of the art genetics research has unveiled a large number of variants linked to ASD. But in general it remains unclear, what biological factors lead to changes in the brains of autistic individuals. We build on the premise that these heterogeneous genetic or genomic aberra- tions will converge towards a common impact downstream, which might be reflected in the transcriptomes of individuals with ASD. Similarly, a considerable number of transcriptome analyses have been performed in attempts to address this question, but their findings lack a clear consensus. As a result, each of these individual studies has not led to any significant advance in understanding the autistic phenotype as a whole. The goal of this research is to comprehensively re-evaluate these expression profiling studies by conducting a systematic meta-analysis. Here, we report a meta-analysis of over 1000 microarrays across twelve independent studies on expression changes in ASD compared to unaffected individuals, in blood and brain. We identified a number of genes that are consistently differentially expressed across studies of the brain, suggestive of effects on mitochondrial function. In blood, consistent changes were more difficult to identify, despite individual studies tending to exhibit larger effects than the brain studies. -

Gene Ontology Supplemental Tables
Gene Ontology Supplemental Tables Summary This file contains the full results for the combined gene set enrichment analysis and the DMR and DMP only enrichment analyses performed with clusterProfiler. DMP enrichment tests also were performed with methylGSA. clusterProfiler DMR & DMP Ontology Description GeneRatio BgRatio pvalue qvalue geneID BP cognition 11/136 284/17397 0.0000 0.038 TTC8/ADORA1/CNTNAP2/MEF2C/MEIS2/NTSR1/PAFAH1B1/ ADGRB3/RASGRF1/SLC6A4/SYNGAP1 BP learning or 9/136 246/17397 0.0001 0.114 CNTNAP2/MEF2C/MEIS2/NTSR1/PAFAH1B1/ADGRB3/RASGRF1/ memory SLC6A4/SYNGAP1 BP detection of 3/136 15/17397 0.0002 0.114 ADORA1/ARRB2/NTSR1 temperature stimulus i BP detection of 3/136 15/17397 0.0002 0.114 ADORA1/ARRB2/NTSR1 temperature stimulus i BP dendrite 8/136 216/17397 0.0003 0.114 CTNND2/COBL/MAP2/MEF2C/PAFAH1B1/ADGRB3/KLF7/SYNGAP1 development BP detection of 3/136 19/17397 0.0004 0.114 ADORA1/ARRB2/NTSR1 temperature stimulus BP modulation of 11/136 417/17397 0.0004 0.114 ADORA1/SYT9/ARRB2/MEF2C/NTSR1/RASGRF1/SLC6A4/TMEM108/ chemical YWHAG/SYNGAP1/CLSTN3 synaptic tra BP regulation of 11/136 418/17397 0.0004 0.114 ADORA1/SYT9/ARRB2/MEF2C/NTSR1/RASGRF1/SLC6A4/TMEM108/ trans-synaptic YWHAG/SYNGAP1/CLSTN3 signal BP platelet 3/136 20/17397 0.0005 0.114 ZFPM1/MEF2C/MYH9 formation BP establishment 6/136 128/17397 0.0005 0.114 SDCCAG8/SH3BP1/MAP2/MYH9/PAFAH1B1/FRMD4A of cell polarity BP actin filament- 15/136 723/17397 0.0005 0.114 ABI2/ADORA1/DIAPH2/COBL/SH3BP1/KCNJ5/MEF2C/MYH9/NEB/ based process PAFAH1B1/PLS3/LURAP1/ACTN4/MYOM2/TBCK BP platelet -

Hearing Function and Thresholds: a Genome-Wide Association Study In
Hearing function and thresholds: a genome-wide association study in European isolated populations identifies new loci and pathways Giorgia Girotto, Nicola Pirastu, Rossella Sorice, Ginevra Biino, Harry Campbell, Adamo P. d’Adamo, Nicholas D. Hastie, Teresa Nutile, Ozren Polasek, Laura Portas, et al. To cite this version: Giorgia Girotto, Nicola Pirastu, Rossella Sorice, Ginevra Biino, Harry Campbell, et al.. Hearing function and thresholds: a genome-wide association study in European isolated populations identifies new loci and pathways. Journal of Medical Genetics, BMJ Publishing Group, 2011, 48 (6), pp.369. 10.1136/jmg.2010.088310. hal-00623287 HAL Id: hal-00623287 https://hal.archives-ouvertes.fr/hal-00623287 Submitted on 14 Sep 2011 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Hearing function and thresholds: a genome-wide association study in European isolated populations identifies new loci and pathways Giorgia Girotto1*, Nicola Pirastu1*, Rossella Sorice2, Ginevra Biino3,7, Harry Campbell6, Adamo P. d’Adamo1, Nicholas D. Hastie4, Teresa