Study Protocol and Statistical Analysis Plan
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

WHO Drug Information Vol. 12, No. 3, 1998
WHO DRUG INFORMATION VOLUME 12 NUMBER 3 • 1998 RECOMMENDED INN LIST 40 INTERNATIONAL NONPROPRIETARY NAMES FOR PHARMACEUTICAL SUBSTANCES WORLD HEALTH ORGANIZATION • GENEVA Volume 12, Number 3, 1998 World Health Organization, Geneva WHO Drug Information Contents Seratrodast and hepatic dysfunction 146 Meloxicam safety similar to other NSAIDs 147 Proxibarbal withdrawn from the market 147 General Policy Issues Cholestin an unapproved drug 147 Vigabatrin and visual defects 147 Starting materials for pharmaceutical products: safety concerns 129 Glycerol contaminated with diethylene glycol 129 ATC/DDD Classification (final) 148 Pharmaceutical excipients: certificates of analysis and vendor qualification 130 ATC/DDD Classification Quality assurance and supply of starting (temporary) 150 materials 132 Implementation of vendor certification 134 Control and safe trade in starting materials Essential Drugs for pharmaceuticals: recommendations 134 WHO Model Formulary: Immunosuppressives, antineoplastics and drugs used in palliative care Reports on Individual Drugs Immunosuppresive drugs 153 Tamoxifen in the prevention and treatment Azathioprine 153 of breast cancer 136 Ciclosporin 154 Selective serotonin re-uptake inhibitors and Cytotoxic drugs 154 withdrawal reactions 136 Asparaginase 157 Triclabendazole and fascioliasis 138 Bleomycin 157 Calcium folinate 157 Chlormethine 158 Current Topics Cisplatin 158 Reverse transcriptase activity in vaccines 140 Cyclophosphamide 158 Consumer protection and herbal remedies 141 Cytarabine 159 Indiscriminate antibiotic -

The Double Stranded RNA Analog Poly-IC Elicits Both Robust IFN-Λ Production and Oncolytic Activity in Human Gastrointestinal Cancer Cells
www.oncotarget.com Oncotarget, 2018, Vol. 9, (No. 77), pp: 34471-34484 Research Paper The double stranded RNA analog poly-IC elicits both robust IFN-λ production and oncolytic activity in human gastrointestinal cancer cells Chantal Bou-Hanna1,*, Anne Jarry1,2,*, Jean-François Mosnier1,3, Céline Bossard1,2,3 and Christian L. Laboisse1,3 1University of Nantes, EA4273 Biometadys, Nantes, France 2Current address: CRCINA, INSERM, Université d’Angers, Université de Nantes, Nantes, France 3Pathology Department, Nantes University Hospital, Nantes, France *Equal co-authors Correspondence to: Christian L. Laboisse, email: [email protected]; [email protected] Keywords: dsRNA/poly-IC; IFN-λ; immunoadjuvant; oncolysis; human gastrointestinal cancer Received: February 09, 2018 Accepted: September 06, 2018 Published: October 02, 2018 Copyright: Bou-Hanna et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License 3.0 (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Purpose: Type III IFN (IFN-λ) is the dominant frontline response over type I IFN in human normal intestinal epithelial cells upon viral infection, this response being mimicked by the dsRNA analog poly-IC. Poly-IC also induces cell death in murine intestinal crypts ex vivo. Here we examined whether these innate defense functions of normal intestinal epithelial cells are recapitulated in gastrointestinal carcinoma cells so that they could be harnessed to exert both immunoadjuvant and oncolytic functions, an unknown issue yet. Experimental design: Four human gastrointestinal carcinoma cell lines versus the Jurkat lymphoma cell line were used to assess the effects of intracellular poly-IC on i) IFN-λ secretion and cell proliferation and ii) role of NFκB signaling using the NFκB inhibitory peptide SN50 as a screening probe and a siRNA approach. -

Patient Resource Free
PATIENT RESOURCE FREE Third Edition CancerUnderstanding Immunotherapy Published in partnership with CONTENT REVIEWED BY A DISTINGUISHED PRP MEDICAL PATIENT ADVISORY RESOURCE BOARD PUBLISHING® Understanding TABLE OF CONTENTS Cancer Immunotherapy Third Edition IN THIS GUIDE 1 Immunotherapy Today 2 The Immune System 4 Immunotherapy Strategies 6 Melanoma Survivor Story: Jane McNee Chief Executive Officer Mark A. Uhlig I didn’t look sick, so I didn’t want to act sick. Publisher Linette Atwood Having and treating cancer is only one part of your life. Co-Editor-in-Chief Charles M. Balch, MD, FACS Jane McNee, melanoma survivor Co-Editor-in-Chief Howard L. Kaufman, MD, FACS Senior Vice President Debby Easum 7 The Road to Immunotherapy Vice President, Operations Leann Sandifar 8 Cancer Types Managing Editor Lori Alexander, MTPW, ELS, MWC™ 14 Side Effects Senior Editors Dana Campbell Colleen Scherer 15 Glossary Graphic Designer Michael St. George 16 About Clinical Trials Medical Illustrator Todd Smith 16 Cancer Immunotherapy Clinical Trials by Disease Production Manager Jennifer Hiltunen 35 Support & Financial Resources Vice Presidents, Amy Galey Business Development Kathy Hungerford 37 Notes Stephanie Myers Kenney Account Executive Melissa Amaya Office Address 8455 Lenexa Drive CO-EDITORS-IN-CHIEF Overland Park, KS 66214 For Additional Information [email protected] Charles M. Balch, MD, FACS Advisory Board Visit our website at Professor of Surgery, The University of Texas PatientResource.com to read bios of MD Anderson Cancer Center our Medical and Patient Advisory Board. Editor-in-Chief, Patient Resource LLC Editor-in-Chief, Annals of Surgical Oncology Past President, Society of Surgical Oncology For Additional Copies: To order additional copies of Patient Resource Cancer Guide: Understanding Cancer Immunotherapy, Howard L. -

Tanibirumab (CUI C3490677) Add to Cart
5/17/2018 NCI Metathesaurus Contains Exact Match Begins With Name Code Property Relationship Source ALL Advanced Search NCIm Version: 201706 Version 2.8 (using LexEVS 6.5) Home | NCIt Hierarchy | Sources | Help Suggest changes to this concept Tanibirumab (CUI C3490677) Add to Cart Table of Contents Terms & Properties Synonym Details Relationships By Source Terms & Properties Concept Unique Identifier (CUI): C3490677 NCI Thesaurus Code: C102877 (see NCI Thesaurus info) Semantic Type: Immunologic Factor Semantic Type: Amino Acid, Peptide, or Protein Semantic Type: Pharmacologic Substance NCIt Definition: A fully human monoclonal antibody targeting the vascular endothelial growth factor receptor 2 (VEGFR2), with potential antiangiogenic activity. Upon administration, tanibirumab specifically binds to VEGFR2, thereby preventing the binding of its ligand VEGF. This may result in the inhibition of tumor angiogenesis and a decrease in tumor nutrient supply. VEGFR2 is a pro-angiogenic growth factor receptor tyrosine kinase expressed by endothelial cells, while VEGF is overexpressed in many tumors and is correlated to tumor progression. PDQ Definition: A fully human monoclonal antibody targeting the vascular endothelial growth factor receptor 2 (VEGFR2), with potential antiangiogenic activity. Upon administration, tanibirumab specifically binds to VEGFR2, thereby preventing the binding of its ligand VEGF. This may result in the inhibition of tumor angiogenesis and a decrease in tumor nutrient supply. VEGFR2 is a pro-angiogenic growth factor receptor -

And Poly-ICLC in Glioblastoma
De Waele et al. Journal of Experimental & Clinical Cancer Research (2021) 40:213 https://doi.org/10.1186/s13046-021-02017-2 REVIEW Open Access A systematic review on poly(I:C) and poly- ICLC in glioblastoma: adjuvants coordinating the unlocking of immunotherapy Jorrit De Waele1* , Tias Verhezen1, Sanne van der Heijden1, Zwi N. Berneman2,3,4, Marc Peeters1,5, Filip Lardon1, An Wouters1† and Evelien L. J. M. Smits1,4† Abstract Immunotherapy is currently under intensive investigation as a potential breakthrough treatment option for glioblastoma. Given the anatomical and immunological complexities surrounding glioblastoma, lymphocytes that infiltrate the brain to develop durable immunity with memory will be key. Polyinosinic:polycytidylic acid, or poly(I:C), and its derivative poly-ICLC could serve as a priming or boosting therapy to unleash lymphocytes and other factors in the (immuno)therapeutic armory against glioblastoma. Here, we present a systematic review on the effects and efficacy of poly(I:C)/poly-ICLC for glioblastoma treatment, ranging from preclinical work on cellular and murine glioblastoma models to reported and ongoing clinical studies. MEDLINE was searched until 15 May 2021 to identify preclinical (glioblastoma cells, murine models) and clinical studies that investigated poly(I:C) or poly-ICLC in glioblastoma. A systematic review approach was conducted according to PRISMA guidelines. ClinicalTrials.gov was queried for ongoing clinical studies. Direct pro-tumorigenic effects of poly(I:C) on glioblastoma cells have not been described. On the contrary, poly(I:C) changes the immunological profile of glioblastoma cells and can also kill them directly. In murine glioblastoma models, poly(I:C) has shown therapeutic relevance as an adjuvant therapy to several treatment modalities, including vaccination and immune checkpoint blockade. -
![Ehealth DSI [Ehdsi V2.2.2-OR] Ehealth DSI – Master Value Set](https://docslib.b-cdn.net/cover/8870/ehealth-dsi-ehdsi-v2-2-2-or-ehealth-dsi-master-value-set-1028870.webp)
Ehealth DSI [Ehdsi V2.2.2-OR] Ehealth DSI – Master Value Set
MTC eHealth DSI [eHDSI v2.2.2-OR] eHealth DSI – Master Value Set Catalogue Responsible : eHDSI Solution Provider PublishDate : Wed Nov 08 16:16:10 CET 2017 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 1 of 490 MTC Table of Contents epSOSActiveIngredient 4 epSOSAdministrativeGender 148 epSOSAdverseEventType 149 epSOSAllergenNoDrugs 150 epSOSBloodGroup 155 epSOSBloodPressure 156 epSOSCodeNoMedication 157 epSOSCodeProb 158 epSOSConfidentiality 159 epSOSCountry 160 epSOSDisplayLabel 167 epSOSDocumentCode 170 epSOSDoseForm 171 epSOSHealthcareProfessionalRoles 184 epSOSIllnessesandDisorders 186 epSOSLanguage 448 epSOSMedicalDevices 458 epSOSNullFavor 461 epSOSPackage 462 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 2 of 490 MTC epSOSPersonalRelationship 464 epSOSPregnancyInformation 466 epSOSProcedures 467 epSOSReactionAllergy 470 epSOSResolutionOutcome 472 epSOSRoleClass 473 epSOSRouteofAdministration 474 epSOSSections 477 epSOSSeverity 478 epSOSSocialHistory 479 epSOSStatusCode 480 epSOSSubstitutionCode 481 epSOSTelecomAddress 482 epSOSTimingEvent 483 epSOSUnits 484 epSOSUnknownInformation 487 epSOSVaccine 488 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 3 of 490 MTC epSOSActiveIngredient epSOSActiveIngredient Value Set ID 1.3.6.1.4.1.12559.11.10.1.3.1.42.24 TRANSLATIONS Code System ID Code System Version Concept Code Description (FSN) 2.16.840.1.113883.6.73 2017-01 A ALIMENTARY TRACT AND METABOLISM 2.16.840.1.113883.6.73 2017-01 -

IFN-Γ- and IL-17-Producing CD8+ T (Tc17-1)
Open access Short report J Immunother Cancer: first published as 10.1136/jitc-2021-002426 on 30 June 2021. Downloaded from IFN-γ- and IL-17- producing CD8+ T (Tc17-1) cells in combination with poly- ICLC and peptide vaccine exhibit antiglioma activity Takayuki Ohkuri,1,2 Akemi Kosaka,1,2 Maki Ikeura,2 Andres M Salazar,3 1,2,4,5 Hideho Okada To cite: Ohkuri T, Kosaka A, ABSTRACT led to Food and Drug Administration Ikeura M, et al. IFN-γ- and IL- Background While adoptive transfer of T- cells has been approvals for patients with B- cell malig- 17- producing CD8+ T (Tc17-1) a major medical breakthrough for patients with B cell nancies. However, the development of cells in combination with poly- malignancies, the development of safe and effective T- ICLC and peptide vaccine exhibit effective T- cell therapies for GBM remains cell- based immunotherapy for central nervous system antiglioma activity. Journal a challenge due to multiple barriers, (CNS) tumors, such as glioblastoma (GBM), still needs to for ImmunoTherapy of Cancer including restricted T- cell homing to GBM overcome multiple challenges, including effective homing 2021;9:e002426. doi:10.1136/ and the antigenic heterogeneity, as we jitc-2021-002426 and persistence of T- cells. Based on previous observations 2 that interleukin (IL)-17-producing T-cells can traffic to the reviewed recently. Especially, the central + nervous system (CNS) has anatomical ► Additional online CNS in autoimmune conditions, we evaluated CD8 T- cells supplemental material is that produce IL-17 and interferon-γ (IFN-γ) (Tc17-1) cells barriers (eg, blood–brain barrier) which 3 published online only. -

BMJ Open Is Committed to Open Peer Review. As Part of This Commitment We Make the Peer Review History of Every Article We Publish Publicly Available
BMJ Open is committed to open peer review. As part of this commitment we make the peer review history of every article we publish publicly available. When an article is published we post the peer reviewers’ comments and the authors’ responses online. We also post the versions of the paper that were used during peer review. These are the versions that the peer review comments apply to. The versions of the paper that follow are the versions that were submitted during the peer review process. They are not the versions of record or the final published versions. They should not be cited or distributed as the published version of this manuscript. BMJ Open is an open access journal and the full, final, typeset and author-corrected version of record of the manuscript is available on our site with no access controls, subscription charges or pay-per-view fees (http://bmjopen.bmj.com). If you have any questions on BMJ Open’s open peer review process please email [email protected] BMJ Open Pediatric drug utilization in the Western Pacific region: Australia, Japan, South Korea, Hong Kong and Taiwan Journal: BMJ Open ManuscriptFor ID peerbmjopen-2019-032426 review only Article Type: Research Date Submitted by the 27-Jun-2019 Author: Complete List of Authors: Brauer, Ruth; University College London, Research Department of Practice and Policy, School of Pharmacy Wong, Ian; University College London, Research Department of Practice and Policy, School of Pharmacy; University of Hong Kong, Centre for Safe Medication Practice and Research, Department -

Delineating the Unique Functions of Ifnα and Ifnβ Context Is
Washington University School of Medicine Digital Commons@Becker Open Access Publications 1-1-2020 Context Is key: Delineating the unique functions of IFNα and IFNβ in disease Lindsey E Fox Marissa C Locke Deborah J Lenschow Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs REVIEW published: 21 December 2020 doi: 10.3389/fimmu.2020.606874 Context Is Key: Delineating the Unique Functions of IFNa and IFNb in Disease Lindsey E. Fox 1†, Marissa C. Locke 1† and Deborah J. Lenschow 1,2* 1 Department of Pathology and Immunology, Washington University School of Medicine, Saint Louis, MO, United States, 2 Department of Medicine, Washington University School of Medicine, Saint Louis, MO, United States Type I interferons (IFNs) are critical effector cytokines of the immune system and were originally known for their important role in protecting against viral infections; however, they have more recently been shown to play protective or detrimental roles in many disease states. Type I IFNs consist of IFNa, IFNb, IFNϵ, IFNk, IFNw, and a few others, and they all signal through a shared receptor to exert a wide range of biological activities, including antiviral, antiproliferative, proapoptotic, and immunomodulatory effects. Though the Edited by: individual type I IFN subtypes possess overlapping functions, there is growing Mark R. Walter, University of Alabama at Birmingham, appreciation that they also have unique properties. In this review, we summarize some United States of the mechanisms underlying differential expression of and signaling by type I IFNs, and Reviewed by: we discuss examples of differential functions of IFNa and IFNb in models of infectious ´ Gilles Uze, disease, cancer, and autoimmunity. -
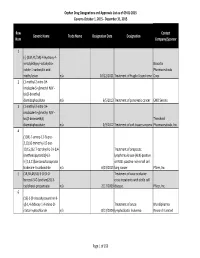
Orphan Drug Designation List
Orphan Drug Designations and Approvals List as of 09‐01‐2015 Governs October 1, 2015 ‐ December 31, 2015 Row Contact Generic Name Trade Name Designation Date Designation Num Company/Sponsor 1 (‐)‐(3aR,4S,7aR)‐4‐Hydroxy‐4‐ m‐tolylethynyl‐octahydro‐ Novartis indole‐1‐carboxylic acid Pharmaceuticals methyl ester n/a 10/12/2011 Treatment of Fragile X syndrome Corp. 2 (1‐methyl‐2‐nitro‐1H‐ imidazole‐5‐yl)methyl N,N'‐ bis(2‐broethyl) diamidophosphate n/a 6/5/2013 Treatment of pancreatic cancer EMD Serono 3 (1‐methyl‐2‐nitro‐1H‐ imidazole‐5‐yl)methyl N,N'‐ bis(2‐bromoethyl) Threshold diamidophosphate n/a 3/9/2012 Treatment of soft tissue sarcoma Pharmaceuticals, Inc. 4 (1OR)‐7‐amino‐12‐fluoro‐ 2,10,16‐trimethyl‐15 oxo‐ 10,15,16,17‐tetrahydro‐2H‐8,4‐ Treatment of anaplastic (metheno)pyrazolo[4,3‐ lymphoma kinase (ALK)‐positive h][2,5,11]benzoxadiazacyclote or ROS1‐positive non‐small cell tradecine‐3‐carbonitrile n/a 6/23/2015 lung cancer Pfizer, Inc. 5 (1R,3R,4R,5S)‐3‐O‐[2‐O‐ Treatment of vaso‐occlusive benzoyl‐3‐O‐(sodium(2S)‐3‐ crisis in patients with sickle cell cyclohexyl‐propanoate‐ n/a 2/17/2009 disease. Pfizer, Inc. 6 (1S)‐1‐(9‐deazahypoxanthin‐9‐ yl)‐1,4‐dideoxy‐1,4‐imino‐D‐ Treatment of acute Mundipharma ribitol‐hydrochloride n/a 8/13/2004 lymphoblastic leukemia Research Limited Page 1 of 359 Orphan Drug Designations and Approvals List as of 09‐01‐2015 Governs October 1, 2015 ‐ December 31, 2015 Row Contact Generic Name Trade Name Designation Date Designation Num Company/Sponsor 7 Treatment of chronic lymphocytic leukemia and related leukemias to include (1S)‐1‐(9‐deazahypoxanthin‐9‐ prolymphocytic leukemia, adult T‐ yl)‐1,4‐dideoxy‐1,4‐imino‐D‐ cell leukemia, and hairy cell Mundipharma ribitol‐hydrochloride n/a 8/10/2004 leukemia Research Ltd. -

© Springer Nature Switzerland AG 2019 D. J. A. Crommelin Et Al. (Eds
Index A Antibiotic agents, 75 Abatacept, 557, 586, 590 Antibody dependent cellular cytotoxicity (ADCC), 30, 160, 604 Absorption, 167–168 Antibody-dependent cellular phagocytosis (ADCP), 160 Absorption, distribution, metabolism, elimination (ADME), 361 Antibody-drug conjugates (ADCs), 166, 489, 501 Absorption rate-limited kinetics, 441 Antibody fragments, 151 Accelerated degradation, 86 Antibody-mediated cytotoxicity, 544 Acceptable Operating Ranges, 79 Antibody-mediated rejection (AMR), 537, 547, 548 Acellular pertussis vaccines, 293 Antibody pharmacodynamics, 151 Actimmune®, 633 Antibody pharmacokinetics, 166–180 Active pharmaceutical ingredient (API), 83 Antibody structure, 151, 158–162 Actual wholesale price (AWP), 550 Anti-CD19, 371 Acute rejection, 544, 550 Anti CD20 antibodies, 491–498 Adalimumab, 557, 585 Anti CTLA4 antibodies, 512 Adaptive immune response, 285 Anti-drug antibodies (ADAs), 139, 159 Adaptive immune system, 281, 370 Anti-EGFR, 502–507 Adeno associated virus (AAV), 370 Anti-EGFR-strategies, 502–507 Adenosine deaminase severe combined immunodeficiency Anti-fibrotic, 368 disease (ADA SCID), 370 Antigen-presenting cells (APCs), 286 Adenovirus (Ad), 292, 370 Antigen processing, 287 Adipocytes, 365 Anti growth factor receptor antibodies, 502–504 Adjuvants, 286 Anti growth factor receptor antibodies anti-EGFR2, 507–508 Ado-Trastuzumab-/Trastuzumab Emtanside, 507 Anti-hGH antibodies, 446 Adsorption, 87, 89, 90 Anti-idiotypic IgE, 544 Adsorption chromatography, 69 Anti-IL-1β, 588 Advanced therapies, 357 Anti-IL-5, 603 Advanced -

Alphabetical Listing of ATC Drugs & Codes
Alphabetical Listing of ATC drugs & codes. Introduction This file is an alphabetical listing of ATC codes as supplied to us in November 1999. It is supplied free as a service to those who care about good medicine use by mSupply support. To get an overview of the ATC system, use the “ATC categories.pdf” document also alvailable from www.msupply.org.nz Thanks to the WHO collaborating centre for Drug Statistics & Methodology, Norway, for supplying the raw data. I have intentionally supplied these files as PDFs so that they are not quite so easily manipulated and redistributed. I am told there is no copyright on the files, but it still seems polite to ask before using other people’s work, so please contact <[email protected]> for permission before asking us for text files. mSupply support also distributes mSupply software for inventory control, which has an inbuilt system for reporting on medicine usage using the ATC system You can download a full working version from www.msupply.org.nz Craig Drown, mSupply Support <[email protected]> April 2000 A (2-benzhydryloxyethyl)diethyl-methylammonium iodide A03AB16 0.3 g O 2-(4-chlorphenoxy)-ethanol D01AE06 4-dimethylaminophenol V03AB27 Abciximab B01AC13 25 mg P Absorbable gelatin sponge B02BC01 Acadesine C01EB13 Acamprosate V03AA03 2 g O Acarbose A10BF01 0.3 g O Acebutolol C07AB04 0.4 g O,P Acebutolol and thiazides C07BB04 Aceclidine S01EB08 Aceclidine, combinations S01EB58 Aceclofenac M01AB16 0.2 g O Acefylline piperazine R03DA09 Acemetacin M01AB11 Acenocoumarol B01AA07 5 mg O Acepromazine N05AA04