RBP-6000 Briefing Document: 31 October 2017
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Opioid Dependence: Buprenorphine Prolonged- Release Injection (Buvidal)
Evidence review Opioid dependence: buprenorphine prolonged- release injection (Buvidal) Publication date: February 2019 This evidence review sets out the best available evidence on buprenorphine prolonged-release injection (Buvidal) for treating opioid dependence. It should be read in conjunction with the evidence summary, which gives the key messages. Commissioned by Public Health England. Disclaimer The content of this evidence review was up-to-date in February 2019. See summaries of product characteristics (SPCs), British national formulary (BNF) or the Medicines and Healthcare products Regulatory Agency (MHRA) or NICE websites for up-to-date information. Copyright © NICE 2019. All rights reserved. Subject to Notice of rights. ISBN: 978-1-4731-3246-7 Evidence review: Opioid dependence: buprenorphine prolonged-release injection (Buvidal) (February 2019) 2 of 29 Contents Contents ..................................................................................................................... 3 Background ................................................................................................................ 4 Product overview ........................................................................................................ 4 Mode of action ........................................................................................................ 4 Regulatory status .................................................................................................... 4 Dosing information ................................................................................................. -

Summary of Investments by Type
COMMON INVESTMENT FUNDS Schedule of Investments March 31, 2017 SUMMARY OF INVESTMENTS BY TYPE Cost Market Value Fixed Income Investments $ $ Short-term investments 42,653,484 42,653,484 Bonds 175,482,352 175,327,122 Mortgage-backed securities 22,199,796 21,785,061 Emerging markets debt 9,619,817 10,899,147 Bank loans - high income fund 20,985,176 23,595,337 Total Fixed Income Investments 270,940,624 274,260,151 Equity-Type Investments Mutual funds Domestic 9,234,353 12,420,750 International 18,849,681 18,688,379 Common stocks Domestic 152,833,551 187,487,257 International 216,167,277 227,850,648 Total Equity-Type Investments 397,084,862 446,447,034 Alternative Investments Funds of hedge funds 38,264,990 46,247,453 Real estate trust fund 6,876,041 10,104,141 Total Alternatives Investments 45,141,031 56,351,594 TOTAL INVESTMENTS 713,166,517 777,058,779 Page 1 of 32 COMMON INVESTMENT FUNDS Schedule of Investments March 31, 2017 SUMMARY OF INVESTMENTS BY FUND Cost Market Value Fixed Income Fund $ $ Short-term investments 13,092,627 13,092,627 Bonds 143,036,345 143,362,214 Mortgage-backed securities 21,372,523 20,977,317 Emerging markets debt 9,619,817 10,899,147 Bank loans - high income fund 20,985,176 23,595,337 208,106,487 211,926,642 Domestic Core Equity Fund Short-term investments 9,127,791 9,127,791 Common stocks 134,983,626 165,021,220 Futures - (5,950) Private placement 4,150 4,150 144,115,567 174,147,211 Small Cap Equity Fund Short-term investments 2,937,066 2,937,066 Mutual funds 9,234,353 12,420,750 Common stocks 17,845,775 22,467,836 -

Medications for Opioid Use Disorder for Healthcare and Addiction Professionals, Policymakers, Patients, and Families
Medications for Opioid Use Disorder For Healthcare and Addiction Professionals, Policymakers, Patients, and Families UPDATED 2020 TREATMENT IMPROVEMENT PROTOCOL TIP 63 Please share your thoughts about this publication by completing a brief online survey at: https://www.surveymonkey.com/r/KAPPFS The survey takes about 7 minutes to complete and is anonymous. Your feedback will help SAMHSA develop future products. TIP 63 MEDICATIONS FOR OPIOID USE DISORDER Treatment Improvement Protocol 63 For Healthcare and Addiction Professionals, Policymakers, Patients, and Families This TIP reviews three Food and Drug Administration-approved medications for opioid use disorder treatment—methadone, naltrexone, and buprenorphine—and the other strategies and services needed to support people in recovery. TIP Navigation Executive Summary For healthcare and addiction professionals, policymakers, patients, and families Part 1: Introduction to Medications for Opioid Use Disorder Treatment For healthcare and addiction professionals, policymakers, patients, and families Part 2: Addressing Opioid Use Disorder in General Medical Settings For healthcare professionals Part 3: Pharmacotherapy for Opioid Use Disorder For healthcare professionals Part 4: Partnering Addiction Treatment Counselors With Clients and Healthcare Professionals For healthcare and addiction professionals Part 5: Resources Related to Medications for Opioid Use Disorder For healthcare and addiction professionals, policymakers, patients, and families MEDICATIONS FOR OPIOID USE DISORDER TIP 63 Contents -

AQR International R.C. Equity Mutual Fund June 30, 2019
AQR International R.C. Equity Mutual Fund June 30, 2019 Portfolio Exposures NAV: $28,103,726 Asset Class Security Description Exposure Quantity Equity 1&1 Drillisch Ord Shs (95,027) (2,846) Equity A P Moller Maersk Ord Shs Class B (320,512) (258) Equity A2A Ord Shs 14,344 8,254 Equity Abn Amro Bank Ord Shs 16,413 766 Equity Acs Actividades De Construccion Y Servicios Ord Shs 102,797 2,571 Equity Adecco Group Ord Shs 124,299 2,066 Equity Adidas N Ord Shs 359,272 1,162 Equity Adyen Ord Shs 222,563 288 Equity Aegon Ord Shs (8,266) (1,658) Equity Aeon Ord Shs (30,924) (1,800) Equity Ageas Ord Shs 50,285 966 Equity Aggreko Ord Shs 20,310 2,020 Equity AGL Energy Ord Shs 10,503 748 Equity Airbus Ord Shs 103,224 727 Equity Aisin Seiki Ord Shs 37,878 1,100 Equity Alfresa Holdings Ord Shs 64,143 2,600 Equity Allianz Ord Shs 659,575 2,732 Equity Alps Alpine Ord Shs 52,252 3,100 Equity Alstom Ord Shs 8,410 181 Equity Altice Europe Ord Shs (55,005) (15,290) Equity Altran Technologies Ord Shs (512,420) (32,244) Equity Amadeus It Group Ord Shs 192,293 2,424 Equity Amcor CDI (12,725) (1,120) Equity AMP Ord Shs 8,312 5,587 Equity ams Ord Shs (13,110) (334) Equity Anglo American Ord Shs 396,321 13,874 Equity Ansell Ord Shs 20,180 1,071 Equity Aristocrat Leisure Ord Shs (9,421) (437) Equity Asahi Intecc Ord Shs (9,853) (400) Equity Asics Ord Shs (15,164) (1,400) Equity ASM International Ord Shs 43,363 665 Equity Asos Ord Shs (129,296) (3,984) Equity ASR Nederland Ord Shs 283,476 6,961 Equity Assicurazioni Generali Ord Shs 47,259 2,506 Equity Astellas Pharma Ord Shs 289,219 20,300 Equity Atlas Copco Ord Shs Class B 8,815 307 Equity Atos Ord Shs 215,926 2,579 Equity Aurizon Holdings Ord Shs 125,889 33,221 rrid-3841199 AQR Capital Management, LLC | Two Greenwich Plaza | Greenwich, CT 06830 | p: +1.203.742.3600 | f: +1.203.742.3100 | w: aqr.com AQR International R.C. -

July 29, 2021 Indivior Announces H1 and Q2 2021 Results; Reiterates Upgraded FY 2021 Guidance; Initiating $100M Share Repurchase Program
July 29, 2021 Indivior Announces H1 and Q2 2021 Results; Reiterates Upgraded FY 2021 Guidance; Initiating $100m Share Repurchase Program Period to June 30th Q2 Q2 H1 H1 2021 2020 2021 2020 $m $m $m $m Net Revenue 201 150 381 303 Operating Profit/(Loss) 73 25 130 (165) Net Income/(Loss) 62 18 142 (145) EPS/(LPS) (cents per share) 8 2 19 (20) Adj. Operating Profit* 66 24 117 26 Adj. Net Income* 49 17 87 14 Adj. EPS* 7 2 12 2 *Adjusted (Adj.) basis excludes the impact of exceptional items as referenced in Note 4. This Release Contains Inside Information. Comment by Mark Crossley, CEO of Indivior PLC “The second quarter saw Indivior make good progress against our Strategic Priorities and deliver strong performance, including our fourth consecutive quarter of double-digit growth from SUBLOCADE® (buprenorphine extended-release) injection. Based on the momentum in the business, we raised our FY 2021 guidance at the end of the quarter. Looking forward, our number one priority continues to be capturing the full transformational value of SUBLOCADE, and it is gratifying to see further uptake of this key asset in targeted Organized Health Systems (OHS), including a $7 million order from a criminal justice system that we believe is pioneering treatment of incarcerated individuals suffering from opioid use disorder (OUD). Additionally, we are seeking to strengthen our leadership position in substance use disorder by securing an exclusive agreement with Aelis Farma for their leading mid-stage asset (P2b) targeting cannabis-related disorders. In light of Indivior’s business outlook in 2021 and beyond, and supported by our strong balance sheet, we will be initiating a $100m share buyback program. -

FTSE Russell Publications
2 FTSE Russell Publications 19 August 2021 FTSE 250 Indicative Index Weight Data as at Closing on 30 June 2021 Index weight Index weight Index weight Constituent Country Constituent Country Constituent Country (%) (%) (%) 3i Infrastructure 0.43 UNITED Bytes Technology Group 0.23 UNITED Edinburgh Investment Trust 0.25 UNITED KINGDOM KINGDOM KINGDOM 4imprint Group 0.18 UNITED C&C Group 0.23 UNITED Edinburgh Worldwide Inv Tst 0.35 UNITED KINGDOM KINGDOM KINGDOM 888 Holdings 0.25 UNITED Cairn Energy 0.17 UNITED Electrocomponents 1.18 UNITED KINGDOM KINGDOM KINGDOM Aberforth Smaller Companies Tst 0.33 UNITED Caledonia Investments 0.25 UNITED Elementis 0.21 UNITED KINGDOM KINGDOM KINGDOM Aggreko 0.51 UNITED Capita 0.15 UNITED Energean 0.21 UNITED KINGDOM KINGDOM KINGDOM Airtel Africa 0.19 UNITED Capital & Counties Properties 0.29 UNITED Essentra 0.23 UNITED KINGDOM KINGDOM KINGDOM AJ Bell 0.31 UNITED Carnival 0.54 UNITED Euromoney Institutional Investor 0.26 UNITED KINGDOM KINGDOM KINGDOM Alliance Trust 0.77 UNITED Centamin 0.27 UNITED European Opportunities Trust 0.19 UNITED KINGDOM KINGDOM KINGDOM Allianz Technology Trust 0.31 UNITED Centrica 0.74 UNITED F&C Investment Trust 1.1 UNITED KINGDOM KINGDOM KINGDOM AO World 0.18 UNITED Chemring Group 0.2 UNITED FDM Group Holdings 0.21 UNITED KINGDOM KINGDOM KINGDOM Apax Global Alpha 0.17 UNITED Chrysalis Investments 0.33 UNITED Ferrexpo 0.3 UNITED KINGDOM KINGDOM KINGDOM Ascential 0.4 UNITED Cineworld Group 0.19 UNITED Fidelity China Special Situations 0.35 UNITED KINGDOM KINGDOM KINGDOM Ashmore -

Morningstar® Developed Markets Ex-North America Target Momentum Indexsm 18 June 2021
Morningstar Indexes | Reconstitution Report Page 1 of 8 Morningstar® Developed Markets ex-North America Target Momentum IndexSM 18 June 2021 The index consists of liquid equities that display above-average return on equity. The indexes also emphasize stocks with increasing fiscal For More Information: earnings estimates and technical price momentum indicators. http://indexes.morningstar.com US: +1 312 384-3735 Europe: +44 20 3194 1082 Reconstituted Holdings Name Ticker Country Sector Rank (WAFFR) Weight (%) KUEHNE & NAGEL INTL AG-REG KNIN Switzerland Industrials 1 0.50 PostNL NV PNL Netherlands Industrials 2 0.50 Uponor Corporation UPONOR Finland Industrials 3 0.51 Smart Metering Systems PLC SMS United Kingdom Industrials 4 0.50 QT GROUP OYJ QTCOM Finland Technology 5 0.50 ASML Holding NV ASML Netherlands Technology 6 0.51 Vectura Group PLC VEC United Kingdom Healthcare 7 0.50 Lasertec Corp 6920 Japan Technology 8 0.52 Troax Group AB Class A TROAX Sweden Industrials 9 0.48 BayCurrent Consulting Inc 6532 Japan Technology 10 0.50 Sagax AB B Shares SAGA B Sweden Real Estate 11 0.50 Bilia AB A BILIa Sweden Consumer Cyclical 12 0.51 Mycronic AB MYCR Sweden Technology 13 0.49 Protector Forsikring ASA PROTCT Norway Financial Services 14 0.49 AP Moller - Maersk AS B MAERSK B Denmark Industrials 15 0.50 Polar Capital Holdings PLC POLR United Kingdom Financial Services 16 0.51 Secunet Security Networks AG YSN Germany Technology 17 0.50 Hermes Intl RMS France Consumer Cyclical 18 0.50 Kety KTY Poland Basic Materials 19 0.51 ASM Intl ASMI Netherlands Technology 20 0.51 Nippon Yusen KK 9101 Japan Industrials 21 0.54 Dexerials Corp. -

United Kingdom Small Company Portfolio-Institutional Class As of July 31, 2021 (Updated Monthly) Source: State Street Holdings Are Subject to Change
United Kingdom Small Company Portfolio-Institutional Class As of July 31, 2021 (Updated Monthly) Source: State Street Holdings are subject to change. The information below represents the portfolio's holdings (excluding cash and cash equivalents) as of the date indicated, and may not be representative of the current or future investments of the portfolio. The information below should not be relied upon by the reader as research or investment advice regarding any security. This listing of portfolio holdings is for informational purposes only and should not be deemed a recommendation to buy the securities. The holdings information below does not constitute an offer to sell or a solicitation of an offer to buy any security. The holdings information has not been audited. By viewing this listing of portfolio holdings, you are agreeing to not redistribute the information and to not misuse this information to the detriment of portfolio shareholders. Misuse of this information includes, but is not limited to, (i) purchasing or selling any securities listed in the portfolio holdings solely in reliance upon this information; (ii) trading against any of the portfolios or (iii) knowingly engaging in any trading practices that are damaging to Dimensional or one of the portfolios. Investors should consider the portfolio's investment objectives, risks, and charges and expenses, which are contained in the Prospectus. Investors should read it carefully before investing. This fund operates as a feeder fund in a master-feeder structure and the holdings listed below are the investment holdings of the corresponding master fund. Your use of this website signifies that you agree to follow and be bound by the terms and conditions of use in the Legal Notices. -
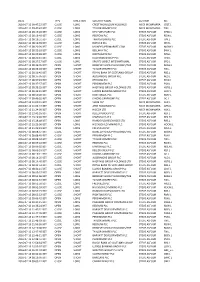
Date Type Direction Security Name Author Ric 2016-07-11
DATE TYPE DIRECTION SECURITY NAME AUTHOR RIC 2016-07-11 09:47:22 BST CLOSE LONG CREST NICHOLSON HOLDINGS MICK MCNAMARA CRST.L 2016-07-11 09:47:47 BST CLOSE LONG TAYLOR WIMPEY PLC MICK MCNAMARA TW.L 2016-07-12 08:19:26 BST CLOSE LONG DFS FURNITURE PLC STEVE ASFOUR DFSD.L 2016-07-12 08:19:40 BST CLOSE LONG REDROW PLC STEVE ASFOUR RDW.L 2016-07-12 08:19:51 BST CLOSE LONG TRAVIS PERKINS PLC STEVE ASFOUR TPK.L 2016-07-12 08:19:59 BST CLOSE LONG IBSTOCK PLC STEVE ASFOUR IBST.L 2016-07-12 08:20:09 BST CLOSE LONG MONEYSUPERMARKET.COM STEVE ASFOUR MONY.L 2016-07-12 08:20:18 BST CLOSE LONG BELLWAY PLC STEVE ASFOUR BWY.L 2016-07-12 08:20:28 BST CLOSE LONG NORTHGATE PLC STEVE ASFOUR NTG.L 2016-07-12 08:20:41 BST CLOSE LONG HALFORDS GROUP PLC STEVE ASFOUR HFD.L 2016-07-12 08:27:17 BST CLOSE LONG SPORTS DIRECT INTERNATIONAL STEVE ASFOUR SPD.L 2016-07-12 08:36:05 BST OPEN SHORT BERKELEY GROUP HOLDINGS/THE STEVE ASFOUR BKGH.L 2016-07-12 08:36:28 BST OPEN SHORT TAYLOR WIMPEY PLC STEVE ASFOUR TW.L 2016-07-12 08:36:45 BST OPEN SHORT ROYAL BANK OF SCOTLAND GROUP STEVE ASFOUR RBS.L 2016-07-12 08:37:05 BST OPEN SHORT ALDERMORE GROUP PLC STEVE ASFOUR ALD.L 2016-07-12 08:37:43 BST OPEN SHORT REDROW PLC STEVE ASFOUR RDW.L 2016-07-12 08:37:57 BST OPEN SHORT PERSIMMON PLC STEVE ASFOUR PSN.L 2016-07-12 08:38:25 BST OPEN SHORT HASTINGS GROUP HOLDINGS LTD STEVE ASFOUR HSTG.L 2016-07-12 08:42:41 BST OPEN SHORT LLOYDS BANKING GROUP PLC STEVE ASFOUR LLOY.L 2016-07-12 08:50:28 BST OPEN SHORT RIGHTMOVE PLC STEVE ASFOUR RMV.L 2016-07-12 09:49:35 BST OPEN SHORT DIXONS CARPHONE -

Transformation
Annual Report 2019 Inspiring patient transformation Annual Report 2019 Contents Inspiring patient Strategic report 1 2019 highlights 3 Chair’s statement 6 Patient stories transformation 10 Chief Executive Officer’s statement and Q&A Delivering on patient needs continues to inspire us 18 Business model at Indivior. We are encouraged that around the world 20 Our stakeholders opioid use disorder is being increasingly recognized 24 Research and development as a treatable medical disease and not a moral failing. 26 Managing our business Ultimately, we believe that patients want, need and responsibly deserve reprieve from the cycle of addiction so that 30 Non-financial information they can recover meaning and purpose to their life. statement 31 Financial review 35 Legal proceedings 39 Risk management 45 Viability statement Governance 46 Chair’s governance statement 48 Board of Directors 50 Executive committee 52 Corporate governance 75 Directors’ remuneration report 92 Directors’ report Financial statements 95 Statement of Directors’ responsibilities 97 Independent Auditor’s report 107 Financial statements 151 Information for shareholders Nathalie’s story p6 Ashlynn’s story p8 2019 highlights Our Vision is that all patients around the world have access to evidence-based treatment for the chronic conditions and co-occurring disorders of addiction. See our business model on pages 18 to 19 2019 Financial Highlights $785m $178m $134m $1,060m Net revenue Operating profit Net income Year-end cash balance (-22% vs. 2018: $1,005m) (-39% vs 2018: $292m) (-51% vs 2018: $275m) (+15% vs 2018: $924m) $72m $202m $176m $821m Net revenue from Adjusted operating profit* Adjusted net income* Year-end net cash balance SUBLOCADE (2018: $12m) (-39% vs 2018: $332m) (-35% vs 2018: $272m) (+21% vs 2018: $681m^) R&D highlights $53m 13 37 R&D investment Peer reviewed Conference (-21% vs. -
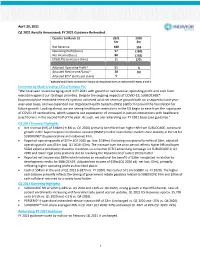
April 29, 2021 Q1 2021 Results Announced; FY 2021 Guidance Reiterated
April 29, 2021 Q1 2021 Results Announced; FY 2021 Guidance Reiterated. Quarter to March 31 2021 2020 $m $m Net Revenue 180 153 Operating Profit/(Loss) 57 (189) Net Income/(Loss) 80 (163) EPS/(LPS) (cents per share) 11 (22) Adjusted Operating Profit* 51 3 Adjusted Net Income/(Loss)* 38 (3) Adjusted EPS* (cents per share) 5 - *Adjusted (Adj.) basis excludes the impact of exceptional items as referenced in Notes 3 and 4. Comment by Mark Crossley, CEO of Indivior PLC “We have seen an encouraging start in FY 2021 with growth in net revenue, operating profit and cash from execution against our strategic priorities. Despite the ongoing impacts of COVID-19, SUBLOCADE® (buprenorphine extended-release) injection achieved solid net revenue growth both on a sequential and year- over-year basis, and we expanded our Organized Health Systems (OHS) platform to build the foundation for future growth. Looking ahead, we are seeing healthcare restrictions in the US begin to ease from the rapid pace of COVID-19 vaccinations, which supports our expectation of increased in-person interactions with healthcare practitioners in the second half of the year. As such, we are reiterating our FY 2021 base case guidance.” Q1 2021 Financial Highlights • Net revenue (NR) of $180m (+18% vs. Q1 2020) primarily benefited from higher NR from SUBLOCADE, continued growth in the buprenorphine medication-assisted (BMAT) market and relative market share stability in the US for SUBOXONE® (buprenorphine and naloxone) Film. • Reported operating profit of $57m (Q1 2020 op. loss: $189m). Excluding exceptional benefits of $6m, adjusted operating profit was $51m (adj. -

M Funds Annual Report 12.31.2020*
Toppan Merrill - SSB_T M Fund_ Inc. Annual Report [Funds] 12-31-2020 ED [AUX] | ltolend | 25-Feb-21 14:27 | 20-39557-2.aa | Sequence: 1 CHKSUM Content: 9591 Layout: 58139 Graphics: 50912 CLEAN M FUND, INC. M International Equity Fund M Large Cap Growth Fund M Capital Appreciation Fund M Large Cap Value Fund Annual Report December 31, 2020 Beginning in February 2021, as permitted by regulations adopted by the Securities and Exchange Commission, paper copies of the Funds’ shareholder reports like this one will no longer be sent by mail, unless you specifically request paper copies of the reports from the Funds or from your financial intermediary, such as a broker-dealer or bank. Instead, the reports will be made available on a website, and you will be notified by mail each time a report is posted and provided with a website link to access the report. If you already elected to receive shareholder reports electronically, you will not be affected by this change and you need not take any action. You may elect to receive shareholder reports and other communications from the Funds electronically by calling your insurance company. If you own these shares through a financial intermediary, you may contact your financial intermediary. You may elect to receive all future reports in paper free of charge. You can inform the Funds that you wish to continue receiving paper copies of your shareholder reports by sending a request in writing to your insurance company or to your financial intermediary. Your election to receive reports in paper will apply to all funds held with the fund complex.