Differential Involvement of Striosome and Matrix Dopamine Systems in a Transgenic Model of Dopa-Responsive Dystonia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Distribution of Tyrosine Hydroxylase-Lmmunoreactive Fibers in Primate Neocortex Is Widespread but Regionally Specific
The Journal of Neuroscience, January 1987, 7(l): 279-290 The Distribution of Tyrosine Hydroxylase-lmmunoreactive Fibers in Primate Neocortex Is Widespread but Regionally Specific David A. Lewis,lea Michael J. Campbell,’ Stephen L. Foote, I.2 Menek Goldstein,3 and John H. Morrison’ ‘Scripps Clinic and Research Foundation, La Jolla, California 92037, 2Department of Psychiatry, University of California, San Diego, California 92037, and 3N.Y.U. Medical Center, New York, New York 10016 An antiserum directed against tyrosine hydroxylase (TH), an alleled by an elaboration and specialization of the noradrenergic enzyme involved in dopamine and norepinephrine synthe- and serotoninergic projections to neocortex. Compared to rat, sis, was used to visualize axons immunohistochemically in both of these systems in primates exhibit substantial regional monkey n’eocortex. Labeled fibers were distributed through- heterogeneity in the density and laminar distribution of cortical out the entire neocottex, but they had striking patterns of fibers (Brown et al., 1979; Morrison et al., 1982a, b; Takeuchi regional and laminar specialization. For example, primary and Sano, 1983). Midbrain dopaminergic neurons also project motor cortex contained the greatest density of TH-labeled to neocortex, but little is known about the distribution of do- fibers, whereas primary sensory regions were sparsely in- paminergic fibers in primate neocortex. Initial studies on the nervated. Marked heterogeneity of fiber density was also rat revealed that dopaminergic fibers were restricted to limited present among the association regions of the frontal, pari- portions of the prefrontal, cingulate, and perirhinal cortical re- etal, and temporal lobes. In addition, the laminar pattern of gions (Thierry et al., 1973; Berger et al., 1974, 1976; Fuxe et innervation in a given region was correlated with its fiber al., 1974; Hokfelt et al., 1974b; Lindvall et al., 1974). -

Insulin-Induced Hypoglycaemia Increases
INSULIN-INDUCED HYPOGLYCAEMIA INCREASES ADRENALINE SECRETION AND TYROSINE HYDROXYLASE PHOSPHORYLATION IN RAT BRAIN AND ADRENAL GLAND L Bobrovskaya, M Senthilkumaran, M Johnson University of South Australia, Sansom Institute for Health Research, Adelaide, SA 5000, Australia [email protected] Introduction Diabetes is Australia’s fastest growing chronic disease. Severe hypoglycaemia is a life- threatening complication of intensive insulin therapy for type 1 and advanced type 2 diabetes. In this study we aimed to determine the effects of insulin-induced hypoglycaemia on adrenaline secretion and tyrosine hydroxylase (TH) phosphorylation (as a marker of cell activation and catecholamine synthesis) in rat brain and adrenal gland. Methods Overnight fasted rats received a single intraperitoneal injection of insulin (10 U/kg) or saline and then were euthanised 30 min, 60 min or 90 min after injection (n=5–6 per group). Cardiac blood was collected for analysis of glucose and adrenaline. Adrenal glands and catecholaminergic brain regions were rapidly removed and TH phosphorylation at Ser40, Ser31, Ser19 and TH protein were analysed by western blotting. Results Blood glucose levels were 6–8 mmol/L in control animals versus 2 mmol/L in insulin-treated animals between 30 and 90 min. Plasma adrenaline (measured by ELISA) was significantly increased at 30 min (20 fold; p<0.05), 60 min (30 fold; p<0.001) and 90 min (13 fold; p<0.001) relative to control. pSer31TH was significantly increased in the adrenal gland at all time points (4–5 fold; p<0.01), in C1 neurons at 30 and 60 min (2 fold; p<0.05), in locus coeruleus and striatum at 60 min (2 fold; p<0.05) and in substantia nigra at 60 at 90 min (2 fold; p<0.01). -

A Dual Role for the Camp-Dependent Protein Kinase in Tyrosine Hydroxylase Gene Expression KWANG-SOO KIM, DONG H
Proc. Natl. Acad. Sci. USA Vol. 90, pp. 3471-3475, April 1993 Neurobiology A dual role for the cAMP-dependent protein kinase in tyrosine hydroxylase gene expression KWANG-SOO KIM, DONG H. PARK, THOMAS C. WESSEL, BAI SONG, JOHN A. WAGNER, AND TONG H. JOH* Laboratory of Molecular Neurobiology, The W. M. Burke Medical Research Institute, Cornell University Medical College, 785 Mamaroneck Avenue, White Plains, NY 10605 Communicated by Erminio Costa, January 11, 1993 ABSTRACT Tyrosine hydroxylase (TH) catalyzes the con- Different signaling pathways are likely to function in post- version of L-tyrosine to 3,4-dihydroxy-L-phenylalanine, the translational and transcriptional responses. For example, first and rate-limiting step in catecholamine biosynthesis. The cAMP-dependent protein kinase (PKA; ATP:protein phos- cAMP-dependent protein kinase (PKA) phosphorylates and photransferase, EC 2.7.1.37) was shown to enhance the activates the TH enzyme and is thought to mediate transcrip- activity of TH, via phosphorylation of the TH protein, by tional induction of the TH gene. To better understand the lowering the Km value for its cofactor, tetrahydrobiopterin (2, functional role of PKA in TH gene regulation, we studied TH 3). Activation ofPKA also appears to induce transcription of gene expression at the transcriptional, translational, and post- the TH gene (8, 9). The 5' flanking sequence of the TH gene translational levels in several PKA-deficient cell lines derived contains several putative cis-acting motifs [e.g., AP1 and AP2 from rat PC12 pheochromocytoma cells. Strikingly, all PKA- binding sites and a cAMP response element (CRE) (10)] deficient cell lines analyzed in this study showed substantial potentially capable of mediating the transcriptional response deficits in basal TH expression as measured by TH enzymatic to the cAMP-mediated PKA signaling pathway. -

Pitfalls in Detection of Contaminating Neuroblastoma Cells by Tyrosine Hydroxylase RT-PCR Due to Catecholamine-Producing Hematopoietic Cells
ANTICANCER RESEARCH 26: 2075-2080 (2006) Pitfalls in Detection of Contaminating Neuroblastoma Cells by Tyrosine Hydroxylase RT-PCR Due to Catecholamine-producing Hematopoietic Cells ZYRAFETE KUÇI, GABRIELE SEITZ, SELIM KUÇI, HERMANN KREYENBERG, MICHAEL SCHUMM, PETER LANG, DIETRICH NIETHAMMER, RUPERT HANDGRETINGER and GERNOT BRUCHELT Department of Hematology and Oncology, Children’s University Hospital, D-72076 Tübingen, Germany Abstract. Background: RT-PCR analysis of compounds of contamination of hematopoietic stem cells by a low number catecholamine metabolism (in particular tyrosine of neuroblastoma cells. hydroxylase, TH) is widely used for the detection of contaminating neuroblastoma cells in hematopoietic stem The contamination of hematopoietic (stem) cells by cell preparations. Due to reports in the literature showing neuroblastoma cells is often analyzed by tyrosine hydroxylase that hematopoietic cells are also able to produce (TH)-RT-PCR, presuming catecholamine metabolism to be a catecholamines, we investigated whether TH-RT-PCR is specific marker for neuroblastoma (1-9). However, two really suitable for this purpose. Materials and Methods: potential problems should be considered with this approach. Besides neuroblastoma cells, mononuclear blood cells, The expression of catecholamine markers in neuroblastoma is apheresis preparations and hematopoietic stem cells were heterogeneous. Some neuroblastoma cells are able to produce used for single and nested RT-PCR. In addition to TH, the dopamine and noradrenaline, some produce only dopamine expressions of dopamine-‚-hydroxylase and noradrenaline and some of them generate only the precursor, DOPA. transporter were analyzed. Results: Using single RT-PCR, a Furthermore, there is also a heterogeneous expression of the clear discrimination between neuroblastoma and hemato- uptake system for catecholamines, dopamine and poietic cells was possible. -

Neural Markers Guide Contents
Neural markers guide Contents Neuroepithelial cells . 4 Radial glia . .5 Immature neurons and intermediate progenitors . 6 Oligodendrocytes and oligodendrocyte precursor cells . .7 Schwann cells and Schwann cell precursors . 8 Astrocytes . 9 Microglia . 10 Mature neurons . 11 Glutamatergic neurons . 12 GABAergic neurons . 13 Dopaminergic neurons . 14 Serotonergic neurons . 15 Cholinergic neurons . 16 References and further reading . 17 Neural lineage markers at a glance . 23 The markers shown in the guide are suggestions based on commonly used markers in published literature . There is often overlap in markers between different cell types, therefore we advise combining multiple markers and observing ultrastructural features where possible . 3 Neuroepithelial cells Neuroepithelial (NE) cells are symmetrically dividing cells that form the neural plate and neural tube during embryonic development . They exhibit typical epithelial features such as tight junctions and are highly polarized along their apical-basal axis . Nestin An intermediate filament protein expressed in NE cells . Its expression persists in radial glia until astrocyte development . Neural progenitor cells derived from human iPSCs stained with anti-nestin (red)(ab105389). SOX2 A transcription factor and the earliest marker of the neural plate . It is expressed in proliferating cells and those that acquire glial fates, but downregulated in post-mitotic neurons . E13 mouse spinal cord sections stained with anti-SOX2 (ab97959). Notch1 A transmembrane receptor that regulates the formation, migration, and differentiation of neural crest cells . Mouse embryo tissue sections stained with anti-notch1 (green) (ab65297). HES1 and HES3 Transcription factors that maintain the symmetrical cell division of NE cells . When NE cells become radial glia, HES3 is downregulated and HES5 is upregulated . -

Increased Activity of Tyrosine Hydroxylase Leads to Elevated Amphetamine Response and Markers of Oxidative Stress in Transgenic Mice
bioRxiv preprint doi: https://doi.org/10.1101/188318; this version posted September 13, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Increased Activity of Tyrosine Hydroxylase Leads to Elevated Amphetamine Response and Markers of Oxidative Stress in Transgenic Mice Laura M. Vecchio1, M. Kristel Bermejo1, Amy Dunn2, Marija Milenkovic1, Nikhil Urs3, Amy Ramsey1, Gary W. Miller2,4,5,6, Ali Salahpour1* 1- Department of Pharmacology and Toxicology, University of Toronto, 2-Department of Environmental Health, Rollins School of Public Health, Emory University, 3-Department of Pharmacology & Therapeutics, University of Florida, 4- Center for Neurodegenerative Diseases, Emory University, 5- Department of Pharmacology, Emory University, 6-Department of Neurology, Emory University. * Corresponding Author: [email protected] Abstract: In Parkinson's disease, noradrenergic cells of the locus coeruleus and dopamine cells within the nigrostriatal pathway undergo profound degeneration. Tyrosine hydroxylase (TH) is the rate-limiting enzyme in the production of all catecholamines, including dopamine and noradrenaline, and is selectively expressed in the cells that produce these neurotransmitters. In vitro studies have previously shown that the TH-synthetic system can contribute to the formation of reactive oxygen species. In addition, animal models of dopamine mishandling demonstrated that free dopamine is neurotoxic. To examine how increased TH activity might influence catecholamine systems in vivo, we generated TH-overexpressing mice (TH-HI) with six total copies of the TH murine gene. A commensurate increase in TH mRNA produced a threefold increase in both total TH protein and phosphorylated TH levels. -

Tyrosine Hydroxylase Deficiency
Tyrosine hydroxylase deficiency Description Tyrosine hydroxylase (TH) deficiency is a disorder that primarily affects movement, with symptoms that may range from mild to severe. The mild form of this disorder is called TH-deficient dopa-responsive dystonia (DRD). Symptoms usually appear during childhood. Affected individuals may exhibit unusual limb positioning and a lack of coordination when walking or running. In some cases, people with TH-deficient DRD have additional movement problems such as shaking when holding a position (postural tremor) or involuntary upward-rolling movements of the eyes. The movement difficulties may slowly increase with age but almost always get better with medical treatment. The severe forms of TH deficiency are called infantile parkinsonism and progressive infantile encephalopathy. These forms of the disorder appear soon after birth and are more difficult to treat effectively. Babies with infantile parkinsonism have delayed development of motor skills such as sitting unsupported or reaching for a toy. They may have stiff muscles, especially in the arms and legs; unusual body positioning; droopy eyelids (ptosis); and involuntary upward-rolling eye movements. The autonomic nervous system, which controls involuntary body functions, may also be affected. Resulting signs and symptoms can include constipation, backflow of stomach acids into the esophagus (gastroesophageal reflux), and difficulty regulating blood sugar, body temperature, and blood pressure. People with the infantile parkinsonism form of the disorder may have intellectual disability, speech problems, attention deficit disorder, and psychiatric conditions such as depression, anxiety, or obsessive-compulsive behaviors. Progressive infantile encephalopathy is an uncommon severe form of TH deficiency. It is characterized by brain dysfunction and structural abnormalities leading to profound physical and intellectual disability. -
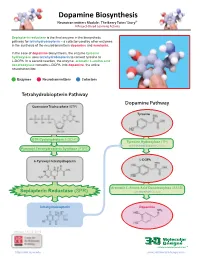
Dopamine Biosynthesis Neurotransmitters Module: the Beery Twins’ Story© a Project-Based Learning Activity
Dopamine Biosynthesis Neurotransmitters Module: The Beery Twins’ Story© A Project-Based Learning Activity Sepiapterin reductase is the final enzyme in the biosynthetic pathway for tetrahydrobiopterin – a cofactor used by other enzymes in the synthesis of the neurotransmitters dopamine and serotonin. In the case of dopamine biosynthesis, the enzyme tyrosine hydroxylase uses tetrahydrobiopterin to convert tyrosine to L-DOPA. In a second reaction, the enzyme aromatic L-amino acid decarboxylase converts L-DOPA into dopamine, the active neurotransmitter. Enzymes Neurotransmitters Cofactors Tetrahydrobiopterin Pathway Dopamine Pathway Guanosine Triphosphate (GTP) Tyrosine GTP Cyclohydrolase I (GCH1) Tyrosine Hydroxylase (TH) with Tetrahydrobiopterin Cofactor Pyruvoyl-Tetrahydropterin Synthase (PTPS) 6-Pyruvoyl-Tetrahydropterin L-DOPA Aromatic L-Amino Acid Decarboxylase (AAAD) Sepiapterin Reductase (SPR) with Vitamin B6 Cofactor Tetrahydrobiopterin Dopamine Version 1.4 -12/2015 ...where molecules become real TM http://cbm.msoe.edu www.3dmoleculardesigns.com Dopamine Biosynthesis Model Guide Neurotransmitters Module: The Beery Twins’ Story© A Project-Based Learning Activity TH AAAD Tyrosine L-DOPA Dopamine Tyrosine (Tyr or Y) is a non-essential amino acid L-DOPA (L-3,4-dihydroxyphenylalanine), an The nal step in the dopamine biosynthesis that can be synthesized in the human body from intermediate molecule in the dopamine biosynthesis pathway requires the removal of the the amino acid phenylalanine. Tyrosine is pathway, is formed by the addition of a hydroxyl carboxylic acid group (COOH) from the composed of the standard amino acid backbone group to the third carbon of the aromatic ring of backbone of the L-DOPA to form the with an aromatic ring containing a hydroxyl (OH) tyrosine. -

Therapeutic Strategies to Treat Or Prevent Off Episodes in Adults With
Drugs https://doi.org/10.1007/s40265-020-01310-2 REVIEW ARTICLE Therapeutic Strategies to Treat or Prevent Of Episodes in Adults with Parkinson’s Disease Nirosen Vijiaratnam1,2 · Thomas Foltynie1,2 © Springer Nature Switzerland AG 2020 Abstract Parkinson’s disease is a chronic, neurodegenerative disease, which manifests with a mixture of motor, cognitive and behav- ioural symptoms. Levodopa is the most efective antiparkinsonian treatment to date, although chronic use engenders a mixture of complications in a substantial proportion of patients. Amongst these is the occurrence of episodes of worsening symptoms—‘of’ phenomena. These episodes can manifest with either motor or non-motor symptoms or a combination of these features and have been found to have profound impacts on patients’ quality of life. Although preventative measures are poorly evidenced, avoiding excessive total daily levodopa intake in selected populations that are deemed to be of a higher risk for developing these episodes warrants further exploration. Methods to improve levodopa bioavailability and delivery to the brain are currently available and are of value in addressing these episodes once they have become established. These include modifcations to levodopa formulations as well as the use of complimentary agents that improve levodopa bioavailability. The deployment of device-assisted approaches is a further dimension that can be considered in addressing these debilitating episodes. This review summarises the clinical manifestations of ‘of’ phenomena and the current approaches to treat them. Although we briefy discuss clinical advances on the horizon, the predominant focus is on existing, established treatments. 1 Introduction Key Points Parkinson’s disease (PD) is the second most highly preva- ‘Of’ phenomena in Parkinson’s disease commonly occur lent neurodegenerative condition and results in substan- as a result of long-term levodopa use and can have a tial patient morbidity and care-giver burden [1]. -

A Guide to Tyrosine Hydroxylase (TH) Deficiency, Or Recessive Dopa- Responsive Dystonia
A Guide to Tyrosine Hydroxylase (TH) Deficiency, or recessive Dopa- responsive Dystonia What is TH Deficiency? Tyrosine hydroxylase (TH) deficiency is a rare metabolic disorder characterized by lack of the enzyme involved in converting the amino acid tyrosine to L-dopa. L-dopa is an important chemical in producing dopamine in the brain. Dopamine is the major neurotransmitter which facilitates motor control and movement. A neurotransmitter is an important chemical messenger that helps nerve cells to communicate properly to each other. TH is a critical enzyme in normal dopamine production, and when it is not working properly to produce enough dopamine, major neurologic abnormalities can occur. In addition, dopamine is also important in making two other important neurotransmitters in the brain and body, norepinephrine (noradrenaline) and epinephrine (adrenaline). When dopamine is critically low, these neurotransmitters may be low too. They play important roles in the brain in regulating attention, and they help to maintain normal blood pressure, body temperature and blood sugar levels. What symptoms are associated with TH deficiency? A wide range of symptoms can be associated with TH deficiency, associated with mild, moderate and severe phenotypes. Mild: In the mildest cases, walking or running may be clumsy but little else may be noticed, at least initially. These symptoms may progress slowly as the child gets older, and may not initially be apparent. Sometimes, one side of the body may seem weaker, or the child may begin to walk up on their tiptoes due to some tightness of the leg muscles. Sometimes these children are diagnosed with cerebral palsy; other times they are simply considered clumsy or uncoordinated. -

The Release of Catecholamines Plays an Essential Role in Initial
0031-3998/02/5102-0207 PEDIATRIC RESEARCH Vol. 51, No. 2, 2002 Copyright © 2002 International Pediatric Research Foundation, Inc. Printed in U.S.A. Long-Term Prenatal Hypoxia Alters Maturation of Adrenal Medulla in Rat JULIE MAMET, JULIE PEYRONNET, JEAN-CHRISTOPHE ROUX, DAVID PERRIN, JEAN-MARIE COTTET-EMARD, JEAN-MARC PEQUIGNOT, HUGO LAGERCRANTZ, AND YVETTE DALMAZ Laboratory of Physiology (Energetic, Cellular, and Molecular Regulation [J.M., J.P., J.-C.R., D.P., J.-M.P., Y.D.], and Laboratory of Environmental Physiology [J.-M.C.-E.], 69373 LYON cedex 08, France; and Department of Woman and Child Health, Karolinska Institute, S-17176 Stockholm, Sweden [J.P., H.L.]. ABSTRACT Catecholamine release from the adrenal medulla glands plays results in major changes in adrenal catecholamine stores and a vital role in postnatal adaptation. A number of pathologic synthesis during the perinatal period, which persist into adult- situations are characterized by oxygen deficiency. The objective hood. The capacity to cope with postnatal stress might be dis- of the present study was to determine the influence of long-term turbed as a consequence of prenatal hypoxia. (Pediatr Res 51: prenatal hypoxia on maturation of the adrenal medulla. Pregnant 207–214, 2002) rats were subjected to hypoxia (10% O2) from the fifth to the 20th d of gestation. The offspring were examined on the 19th d of gestation (E19), the day of birth (P0), and at postnatal (P) day Abbreviations of life P3, P7, P14, P21, and P68. The catecholamine content and DOPA, L-3,4-dihydroxyphenylalanine activity of tyrosine hydroxylase (TH) in vivo were assayed by E, epinephrine HPLC with electrochemical detection. -

Astrocyte-Specific Expression of Tyrosine Hydroxylase After
Gene Therapy (1998) 5, 1650–1655 1998 Stockton Press All rights reserved 0969-7128/98 $12.00 http://www.stockton-press.co.uk/gt Astrocyte-specific expression of tyrosine hydroxylase after intracerebral gene transfer induces behavioral recovery in experimental Parkinsonism J Segovia1, P Vergara1 and M Brenner2 1Departamento de Fisiologı´a, Biofı´sica y Neurociencias, Centro de Investigacio´n y de Estudios Avanzados del IPN, Me´xico; and 2Stroke Branch, National Institute of Neurological Disorders and Stroke, NIH, Bethesda, MD, USA Parkinson’s disease is a neurodegenerative disorder in a rat model of Parkinson’s disease produced by lesion- characterized by the depletion of dopamine in the caudate ing the striatum with 6-hydroxydopamine. Following putamen. Dopamine replacement with levodopa, a precur- microinjection of the transgene into the denervated stria- sor of the neurotransmitter, is presently the most common tum as a DNA–liposome complex, expression of the trans- treatment for this disease. However, in an effort to obtain gene was detected by RT-PCR and TH protein was better therapeutic results, tissue or cells that synthesize observed specifically in astrocytes by using double-labeling catecholamines have been grafted into experimental ani- immunofluorescence for GFAP and TH coupled with laser mals and human patients. In this paper, we present a novel confocal microscopy. Efficacy was demonstrated by sig- technique to express tyrosine hydroxylase (TH) in the nificant behavioral recovery, as assessed by a decrease in host’s own astrocytes. This procedure uses a transgene in the pharmacologically induced turning behavior generated which the expression of a TH cDNA is under the control of by the unilateral denervation of the rat striatum.