LINUX Command Line Reference for Bioinformatics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Installation Guide
install_guide.book Page i Monday, May 11, 2015 8:48 PM Installation Guide Installation Guide Schrödinger Software Release 2015-2 Schrödinger Press install_guide.book Page ii Monday, May 11, 2015 8:48 PM Installation Guide Copyright © 2015 Schrödinger, LLC. All rights reserved. While care has been taken in the preparation of this publication, Schrödinger assumes no responsibility for errors or omissions, or for damages resulting from the use of the information contained herein. Canvas, CombiGlide, ConfGen, Epik, Glide, Impact, Jaguar, Liaison, LigPrep, Maestro, Phase, Prime, PrimeX, QikProp, QikFit, QikSim, QSite, SiteMap, Strike, and WaterMap are trademarks of Schrödinger, LLC. Schrödinger, BioLuminate, and MacroModel are registered trademarks of Schrödinger, LLC. MCPRO is a trademark of William L. Jorgensen. DESMOND is a trademark of D. E. Shaw Research, LLC. Desmond is used with the permission of D. E. Shaw Research. All rights reserved. This publication may contain the trademarks of other companies. Schrödinger software includes software and libraries provided by third parties. For details of the copyrights, and terms and conditions associated with such included third party software, use your browser to open third_party_legal.html, which is in the docs folder of your Schrödinger software installation. This publication may refer to other third party software not included in or with Schrödinger software ("such other third party software"), and provide links to third party Web sites ("linked sites"). References to such other third party software or linked sites do not constitute an endorsement by Schrödinger, LLC or its affiliates. Use of such other third party software and linked sites may be subject to third party license agreements and fees. -

Tortoisemerge a Diff/Merge Tool for Windows Version 1.11
TortoiseMerge A diff/merge tool for Windows Version 1.11 Stefan Küng Lübbe Onken Simon Large TortoiseMerge: A diff/merge tool for Windows: Version 1.11 by Stefan Küng, Lübbe Onken, and Simon Large Publication date 2018/09/22 18:28:22 (r28377) Table of Contents Preface ........................................................................................................................................ vi 1. TortoiseMerge is free! ....................................................................................................... vi 2. Acknowledgments ............................................................................................................. vi 1. Introduction .............................................................................................................................. 1 1.1. Overview ....................................................................................................................... 1 1.2. TortoiseMerge's History .................................................................................................... 1 2. Basic Concepts .......................................................................................................................... 3 2.1. Viewing and Merging Differences ...................................................................................... 3 2.2. Editing Conflicts ............................................................................................................. 3 2.3. Applying Patches ........................................................................................................... -

Programs, Processes and Attacks Windows Live Response/Analysis
Live response Programs, processes and attacks Windows Live response/analysis 101 Linux Live response/analysis 101 Live response data analysis Programs • A compiled Windows program - Portable Executable File format (also called the PE/COFF format) When started certain (imported) DLLs are loaded that is needed by the executable PEview Link Libraries and OS relocation 1 • A dynamic link library (or shared library) takes the idea of an ordinary library (also called a statically linked library) one step further • A dynamic/shared link library is a lot like a program, but instead of being run by the user to do one thing it has a lot of functions "exported" so that other programs can call them – This list, called the export table, gives the address inside the DLL file of each of the functions which the DLL allows other programs to access • The calling executable have a list of imports or imported functions from every DLL file it uses • When Windows loads your program it creates a whole new "address space" for the program • When your program contains the instruction "read memory from address 0x40A0F0 (or something like that) the computer hardware actually looks up in a table to figure out where in physical memory that location is – The address 0x40A0F0 in another program would mean a completely different part of the physical memory of the computer Link Libraries and OS relocation 2 • Programs, when they are loaded, are "mapped" into address space. This process basically copies the code and static data of your program from the executable file -

State Notation Language and Sequencer Users' Guide
State Notation Language and Sequencer Users’ Guide Release 2.0.99 William Lupton ([email protected]) Benjamin Franksen ([email protected]) June 16, 2010 CONTENTS 1 Introduction 3 1.1 About.................................................3 1.2 Acknowledgements.........................................3 1.3 Copyright...............................................3 1.4 Note on Versions...........................................4 1.5 Notes on Release 2.1.........................................4 1.6 Notes on Releases 2.0.0 to 2.0.12..................................7 1.7 Notes on Release 2.0.........................................9 1.8 Notes on Release 1.9......................................... 12 2 Installation 13 2.1 Prerequisites............................................. 13 2.2 Download............................................... 13 2.3 Unpack................................................ 14 2.4 Configure and Build......................................... 14 2.5 Building the Manual......................................... 14 2.6 Test.................................................. 15 2.7 Use.................................................. 16 2.8 Report Bugs............................................. 16 2.9 Contribute.............................................. 16 3 Tutorial 19 3.1 The State Transition Diagram.................................... 19 3.2 Elements of the State Notation Language.............................. 19 3.3 A Complete State Program...................................... 20 3.4 Adding a -

A Rapid, Sensitive, Scalable Method for Precision Run-On Sequencing
bioRxiv preprint doi: https://doi.org/10.1101/2020.05.18.102277; this version posted May 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 A rapid, sensitive, scalable method for 2 Precision Run-On sequencing (PRO-seq) 3 4 Julius Judd1*, Luke A. Wojenski2*, Lauren M. Wainman2*, Nathaniel D. Tippens1, Edward J. 5 Rice3, Alexis Dziubek1,2, Geno J. Villafano2, Erin M. Wissink1, Philip Versluis1, Lina Bagepalli1, 6 Sagar R. Shah1, Dig B. Mahat1#a, Jacob M. Tome1#b, Charles G. Danko3,4, John T. Lis1†, 7 Leighton J. Core2† 8 9 *Equal contribution 10 1Department of Molecular Biology and Genetics, Cornell University, Ithaca, NY 14853, USA 11 2Department of Molecular and Cell Biology, Institute of Systems Genomics, University of 12 Connecticut, Storrs, CT 06269, USA 13 3Baker Institute for Animal Health, College of Veterinary Medicine, Cornell University, Ithaca, 14 NY 14853, USA 15 4Department of Biomedical Sciences, College of Veterinary Medicine, Cornell University, 16 Ithaca, NY 14853, USA 17 #aPresent address: Koch Institute for Integrative Cancer Research, Massachusetts Institute of 18 Technology, Cambridge, MA 02139, USA 19 #bPresent address: Department of Genome Sciences, University of Washington, Seattle, WA, 20 USA 21 †Correspondence to: [email protected] or [email protected] 22 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.05.18.102277; this version posted May 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Specification of the "Sequ" Command
1 Specification of the "sequ" command 2 Copyright © 2013 Bart Massey 3 Revision 0, 1 October 2013 4 This specification describes the "universal sequence" command sequ. The sequ command is a 5 backward-compatible set of extensions to the UNIX [seq] 6 (http://www.gnu.org/software/coreutils/manual/html_node/seq-invo 7 cation.html) command. There are many implementations of seq out there: this 8 specification is built on the seq supplied with GNU Coreutils version 8.21. 9 The seq command emits a monotonically increasing sequence of numbers. It is most commonly 10 used in shell scripting: 11 TOTAL=0 12 for i in `seq 1 10` 13 do 14 TOTAL=`expr $i + $TOTAL` 15 done 16 echo $TOTAL 17 prints 55 on standard output. The full sequ command does this basic counting operation, plus 18 much more. 19 This specification of sequ is in several stages, known as compliance levels. Each compliance 20 level adds required functionality to the sequ specification. Level 1 compliance is equivalent to 21 the Coreutils seq command. 22 The usual specification language applies to this document: MAY, SHOULD, MUST (and their 23 negations) are used in the standard fashion. 24 Wherever the specification indicates an error, a conforming sequ implementation MUST 25 immediately issue appropriate error message specific to the problem. The implementation then 26 MUST exit, with a status indicating failure to the invoking process or system. On UNIX systems, 27 the error MUST be indicated by exiting with status code 1. 28 When a conforming sequ implementation successfully completes its output, it MUST 29 immediately exit, with a status indicating success to the invoking process or systems. -

RSA Adaptive Authentication (On-Premise) 7.3 Operations Guide
RSA® Adaptive Authentication (On-Premise) 7.3 Operations Guide Contact Information Go to the RSA corporate website for regional Customer Support telephone and fax numbers: www.emc.com/domains/rsa/index.htm Trademarks RSA, the RSA Logo, eFraudNetwork and EMC are either registered trademarks or trademarks of EMC Corporation in the United States and/or other countries. All other trademarks used herein are the property of their respective owners. For a list of RSA trademarks, go to www.emc.com/legal/emc-corporation-trademarks.htm#rsa. License Agreement This software and the associated documentation are proprietary and confidential to EMC, are furnished under license, and may be used and copied only in accordance with the terms of such license and with the inclusion of the copyright notice below. This software and the documentation, and any copies thereof, may not be provided or otherwise made available to any other person. No title to or ownership of the software or documentation or any intellectual property rights thereto is hereby transferred. Any unauthorized use or reproduction of this software and the documentation may be subject to civil and/or criminal liability. This software is subject to change without notice and should not be construed as a commitment by EMC. Note on Encryption Technologies This product may contain encryption technology. Many countries prohibit or restrict the use, import, or export of encryption technologies, and current use, import, and export regulations should be followed when using, importing or exporting this product. Distribution Use, copying, and distribution of any EMC software described in this publication requires an applicable software license. -

Transcriptome-Wide M a Detection with DART-Seq
Transcriptome-wide m6A detection with DART-Seq Kate D. Meyer ( [email protected] ) Duke University School of Medicine https://orcid.org/0000-0001-7197-4054 Method Article Keywords: RNA, epitranscriptome, RNA methylation, m6A, methyladenosine, DART-Seq Posted Date: September 23rd, 2019 DOI: https://doi.org/10.21203/rs.2.12189/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/7 Abstract m6A is the most abundant internal mRNA modication and plays diverse roles in gene expression regulation. Much of our current knowledge about m6A has been driven by recent advances in the ability to detect this mark transcriptome-wide. Antibody-based approaches have been the method of choice for global m6A mapping studies. These methods rely on m6A antibodies to immunoprecipitate methylated RNAs, followed by next-generation sequencing to identify m6A-containing transcripts1,2. While these methods enabled the rst identication of m6A sites transcriptome-wide and have dramatically improved our ability to study m6A, they suffer from several limitations. These include requirements for high amounts of input RNA, costly and time-consuming library preparation, high variability across studies, and m6A antibody cross-reactivity with other modications. Here, we describe DART-Seq (deamination adjacent to RNA modication targets), an antibody-free method for global m6A detection. In DART-Seq, the C to U deaminating enzyme, APOBEC1, is fused to the m6A-binding YTH domain. This fusion protein is then introduced to cellular RNA either through overexpression in cells or with in vitro assays, and subsequent deamination of m6A-adjacent cytidines is then detected by RNA sequencing to identify m6A sites. -

Installing MQ 9.1.0.0 LTS to Coexist with MQ 8.0.0.9 in Windows, Including Applying Fix Packs 8.0.0.14 and 9.1.0.4
Page 1 of 47 Installing MQ 9.1.0.0 LTS to coexist with MQ 8.0.0.9 in Windows, including applying Fix Packs 8.0.0.14 and 9.1.0.4 https://www.ibm.com/support/pages/node/2800929 Date last updated: 17-Apr-2020 Angel Rivera – [email protected] IBM MQ Support +++ Objective This techdoc shows all the steps to install MQ 9.1.0.0 LTS in Windows, while co-existing ("side-by-side") with MQ 8.0.0.9. Furthermore, the steps include the installation of the Fix Packs 8.0.0.14 and 9.1.0.4. Note: The particular fix packs mentioned in this tutorial were the latest ones at the time the tutorial was written – the overall recommendation is to apply the most recent fix pack level. None of the installations are designated as “Primary”. Therefore, you must use “setmqenv” to setup the MQ environment for the desired installation. NOTE regarding MQ 9.0: The principles discussed in this techdoc apply also to MQ 9.0. Keep in mind that the End Of Support (EOS) for MQ 8.0 is 30-Apr-2020 and for MQ 9.0 is 30-Sep-2021. At the time this tutorial was written, the EOS for MQ 9.1 has not been announced yet. An important aspect of this feature of multiple installations of MQ (multi-install) in the same host, is that this type of installation does not require that the MQ 8.0 queue managers and applications need to be stopped when doing activities with MQ 9.1. -

Matthew Kendall
Stellaris Toolchain - Matthew Kendall http://www.matthewkendall.com/freesoftware/stellaris-to... Matthew Kendall Search this site Navigation Free Software > Home Stellaris Toolchain Contact Electronics Free Software Notes on how to set up the toolchain for development in C on Luminary Micro's Stellaris ARM Cortex M3 microcontrollers. Photos StellarisWare StellarisWare is Luminary Micro's name for the useful set of libraries that they supply. DriverLib is the Peripheral Driver Library for the on-chip peripherals (and appears to have previously been used as the name of the package as a whole). GrLib is the Graphics Library that provides a set of graphics primitives and a widget set for creating graphical user interfaces on boards that have a graphical display. There is also a bootloader, some (target) utilities and (host) tools. StellarisWare is supplied with evaluation kits and also downloadable from Luminary Micro. Download the version associated with the target board you have to get appropriate example programs. GCC, binutils, et al CodeSourcery says "CodeSourcery, in partnership with ARM, Ltd., develops improvements to the GNU Toolchain for ARM processors and provides regular, validated releases of the GNU Toolchain". CodeSourcery also develops proprietary software, principally startup code and various libraries for use on the target, and a debug "sprite" for use on the development host. This is all bundled together in a package named Sourcery G++ which is available in Personal and Professional editions. Ignore this and download the Lite edition which contains just the 1 de 5 14/05/13 20:45 Stellaris Toolchain - Matthew Kendall http://www.matthewkendall.com/freesoftware/stellaris-to.. -

Introduction to Shell Programming Using Bash Part I
Introduction to shell programming using bash Part I Deniz Savas and Michael Griffiths 2005-2011 Corporate Information and Computing Services The University of Sheffield Email [email protected] [email protected] Presentation Outline • Introduction • Why use shell programs • Basics of shell programming • Using variables and parameters • User Input during shell script execution • Arithmetical operations on shell variables • Aliases • Debugging shell scripts • References Introduction • What is ‘shell’ ? • Why write shell programs? • Types of shell What is ‘shell’ ? • Provides an Interface to the UNIX Operating System • It is a command interpreter – Built on top of the kernel – Enables users to run services provided by the UNIX OS • In its simplest form a series of commands in a file is a shell program that saves having to retype commands to perform common tasks. • Shell provides a secure interface between the user and the ‘kernel’ of the operating system. Why write shell programs? • Run tasks customised for different systems. Variety of environment variables such as the operating system version and type can be detected within a script and necessary action taken to enable correct operation of a program. • Create the primary user interface for a variety of programming tasks. For example- to start up a package with a selection of options. • Write programs for controlling the routinely performed jobs run on a system. For example- to take backups when the system is idle. • Write job scripts for submission to a job-scheduler such as the sun- grid-engine. For example- to run your own programs in batch mode. Types of Unix shells • sh Bourne Shell (Original Shell) (Steven Bourne of AT&T) • csh C-Shell (C-like Syntax)(Bill Joy of Univ. -
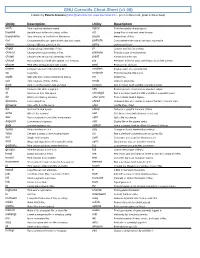
GNU Coreutils Cheat Sheet (V1.00) Created by Peteris Krumins ([email protected], -- Good Coders Code, Great Coders Reuse)
GNU Coreutils Cheat Sheet (v1.00) Created by Peteris Krumins ([email protected], www.catonmat.net -- good coders code, great coders reuse) Utility Description Utility Description arch Print machine hardware name nproc Print the number of processors base64 Base64 encode/decode strings or files od Dump files in octal and other formats basename Strip directory and suffix from file names paste Merge lines of files cat Concatenate files and print on the standard output pathchk Check whether file names are valid or portable chcon Change SELinux context of file pinky Lightweight finger chgrp Change group ownership of files pr Convert text files for printing chmod Change permission modes of files printenv Print all or part of environment chown Change user and group ownership of files printf Format and print data chroot Run command or shell with special root directory ptx Permuted index for GNU, with keywords in their context cksum Print CRC checksum and byte counts pwd Print current directory comm Compare two sorted files line by line readlink Display value of a symbolic link cp Copy files realpath Print the resolved file name csplit Split a file into context-determined pieces rm Delete files cut Remove parts of lines of files rmdir Remove directories date Print or set the system date and time runcon Run command with specified security context dd Convert a file while copying it seq Print sequence of numbers to standard output df Summarize free disk space setuidgid Run a command with the UID and GID of a specified user dir Briefly list directory