An Autosomal Or X Linked Mutation Results in True Hermaphrodites And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Webbed Penis
Kathmandu University Medical Journal (2010), Vol. 8, No. 1, Issue 29, 95-96 Case Note Webbed penis: A rare case Agrawal R1, Chaurasia D2, Jain M3 1Resident in Surgery, 2Associate Professor, Department of Urology, 3Assistant Professor, Department of Plastic and Reconstructive Surgery, MLN Medical College, Allahabad (India) Abstract Webbed penis belongs to a rare and little-known defect of the external genitalia. The term denotes the penis of normal size for age hidden in the adjacent scrotal and pubic tissues. Though rare, it can be treated easily by surgery. A case of webbed penis is presented with brief review of literature. Key words: penis, webbed ebbed penis is a rare anomaly of structure of Wpenis. Though a congenital anomaly, usually the patient presents in late childhood or adolescence. Skin of penis forms the shape of a web, covering whole or part of penis circumferentially; with or without glans, burying the penile tissue inside. The length of shaft is normal with normal stretched length. Phimosis may be present. The penis appears small without any diffi culty in voiding function. Fig 1: Penis showing web Fig 2: Markings for double of skin on anterior Z-plasty on penis Case report aspect Our patient, a 17 year old male, presented to us with congenital webbed penis. On examination, skin webs Discussion were present on both lateral sides from prepuce to lateral Webbed penis is a developmental malformation with aspect of penis.[Fig. 1] On ventral aspect, the skin web less than 60 cases reported in literature. The term was present from prepuce to inferior margin of median denotes the penis of normal size for age hidden in the raphe of scrotum. -
![Springer MRW: [AU:0, IDX:0]](https://docslib.b-cdn.net/cover/3905/springer-mrw-au-0-idx-0-323905.webp)
Springer MRW: [AU:0, IDX:0]
Pre-Testicular, Testicular, and Post- Testicular Causes of Male Infertility Fotios Dimitriadis, George Adonakis, Apostolos Kaponis, Charalampos Mamoulakis, Atsushi Takenaka, and Nikolaos Sofikitis Abstract Infertility is both a private and a social health problem that can be observed in 12–15% of all sexually active couples. The male factor can be diagnosed in 50% of these cases either alone or in combination with a female component. The causes of male infertility can be identified as factors acting at pre-testicular, testicular or post-testicular level. However, despite advancements, predominantly in the genetics of fertility, etiological factors of male infertility cannot be identi- fied in approximately 50% of the cases, classified as idiopathic infertility. On the other hand, the majority of the causes leading to male infertility can be treated or prevented. Thus a full understanding of these conditions is crucial in order to allow the clinical andrologist not simply to retrieve sperm for assisted reproduc- tive techniques purposes, but also to optimize the male’s fertility potential in order to offer the couple the possibility of a spontaneous conceivement. This chapter offers the clinical andrologist a wide overview of pre-testicular, testicular, and post-testicular causes of male infertility. F. Dimitriadis Department of Urology, School of Medicine, Aristotle University, Thessaloniki, Greece e-mail: [email protected] G. Adonakis • A. Kaponis Department of Ob/Gyn, School of Medicine, Patras University, Patras, Greece C. Mamoulakis Department of Urology, School of Medicine, University of Crete, Crete, Greece A. Takenaka Department of Urology, School of Medicine, Tottori University, Yonago, Japan N. Sofikitis (*) Department of Urology, School of Medicine, Ioannina University, Ioannina, Greece e-mail: [email protected] # Springer International Publishing AG 2017 1 M. -

Evaluation of Additional Anomalies in Concomitance of Hypospadias And
Türkiye Çocuk Hastalıkları Dergisi 222 Özgün Araştırma Original Article Turkish Journal of Pediatric Disease Evaluation of Additional Anomalies in Concomitance of Hypospadias and Undescended Testes Hipospadias ve İnmemiş Testis Birlikteliğinde Ek Anomali Sıklığının Değerlendirilmesi Ufuk ATES, Gülnur GÖLLÜ, Nil YAŞAM TAŞTEKİN, Anar QURBANOV, Günay EKBERLİ, Meltem BİNGÖL KOLOĞLU, Emin AYDIN YAĞMURLU, Tanju AKTUĞ, Hüseyin DİNDAR, Ahmet Murat ÇAKMAK Ankara University Medical School, Pediatric Surgery Department, Pediatric Urology Division, Ankara, Turkey ABSTRACT Objective: Hypospadias is a common genitourinary system (GUS) anomaly in boys occurring in 1 of 200 to 300 live births. Undescended testes is frequently detected among accompanying anomalies in cases with hypospadias. Especially in proximal hypospadias and bilateral cases, this association may indicate sexual differentiation disorders. The aim of the study was to evaluate the togetherness of additional anomalies in hypospadiac children with undescended testes. Material and Methods: Between 2007 and 2016, data of 392 children who underwent surgery for hypospadias were evaluated retrospectively. Urethral meatus was present at scrotal and penoscrotal in 65 cases (16.6%) and glanular, coronal, subcoronal and midpenile in 327 cases (83.4%). The cases were divided into two groups as those with both testes in the scrotum and those with undescended testes, and the anomalies were recorded. Results: The mean age of the children with proximal hypospadias was 21 months (6-240 months). Of the children with proximal hypospadias, 26 (40%) had undescended testes and 39 (60%) had testes in the scrotum. Undescended testes were detected bilaterally in 17 patients (65.4%) and unilaterally in nine patients (34.6%) in the undescended testes group. -

Penile Anomalies in Adolescence
Review Special Issue: Penile Anomalies in Children TheScientificWorldJOURNAL (2011) 11, 614–623 TSW Urology ISSN 1537-744X; DOI 10.1100/tsw.2011.38 Penile Anomalies in Adolescence Dan Wood* and Christopher Woodhouse Adolescent Urology Department, University College London Hospitals E-mail: [email protected]; [email protected] Received August 13, 2010; Revised January 9, 2011; Accepted January 11, 2011; Published March 7, 2011 This article considers the impact and outcomes of both treatment and underlying condition of penile anomalies in adolescent males. Major congenital anomalies (such as exstrophy/epispadias) are discussed, including the psychological outcomes, common problems (such as corporal asymmetry, chordee, and scarring) in this group, and surgical assessment for potential surgical candidates. The emergence of new surgical techniques continues to improve outcomes and potentially raises patient expectations. The importance of balanced discussion in conditions such as micropenis, including multidisciplinary support for patients, is important in order to achieve appropriate treatment decisions. Topical treatments may be of value, but in extreme cases, phalloplasty is a valuable option for patients to consider. In buried penis, the importance of careful assessment and, for the majority, a delay in surgery until puberty has completed is emphasised. In hypospadias patients, the variety of surgical procedures has complicated assessment of outcomes. It appears that true surgical success may be difficult to measure as many men who have had earlier operations are not reassessed in either puberty or adult life. There is also a brief discussion of acquired penile anomalies, including causation and treatment of lymphoedema, penile fracture/trauma, and priapism. -

EAU-Guidelines-On-Paediatric-Urology-2019.Pdf
EAU Guidelines on Paediatric Urology C. Radmayr (Chair), G. Bogaert, H.S. Dogan, R. Kocvara˘ , J.M. Nijman (Vice-chair), R. Stein, S. Tekgül Guidelines Associates: L.A. ‘t Hoen, J. Quaedackers, M.S. Silay, S. Undre European Society for Paediatric Urology © European Association of Urology 2019 TABLE OF CONTENTS PAGE 1. INTRODUCTION 8 1.1 Aim 8 1.2 Panel composition 8 1.3 Available publications 8 1.4 Publication history 8 1.5 Summary of changes 8 1.5.1 New and changed recommendations 9 2. METHODS 9 2.1 Introduction 9 2.2 Peer review 9 2.3 Future goals 9 3. THE GUIDELINE 10 3.1 Phimosis 10 3.1.1 Epidemiology, aetiology and pathophysiology 10 3.1.2 Classification systems 10 3.1.3 Diagnostic evaluation 10 3.1.4 Management 10 3.1.5 Follow-up 11 3.1.6 Summary of evidence and recommendations for the management of phimosis 11 3.2 Management of undescended testes 11 3.2.1 Background 11 3.2.2 Classification 11 3.2.2.1 Palpable testes 12 3.2.2.2 Non-palpable testes 12 3.2.3 Diagnostic evaluation 13 3.2.3.1 History 13 3.2.3.2 Physical examination 13 3.2.3.3 Imaging studies 13 3.2.4 Management 13 3.2.4.1 Medical therapy 13 3.2.4.1.1 Medical therapy for testicular descent 13 3.2.4.1.2 Medical therapy for fertility potential 14 3.2.4.2 Surgical therapy 14 3.2.4.2.1 Palpable testes 14 3.2.4.2.1.1 Inguinal orchidopexy 14 3.2.4.2.1.2 Scrotal orchidopexy 15 3.2.4.2.2 Non-palpable testes 15 3.2.4.2.3 Complications of surgical therapy 15 3.2.4.2.4 Surgical therapy for undescended testes after puberty 15 3.2.5 Undescended testes and fertility 16 3.2.6 Undescended -

Experimentally Induced Testicular Dysgenesis Syndrome Originates in the Masculinization Programming Window
Experimentally induced testicular dysgenesis syndrome originates in the masculinization programming window Sander van den Driesche, … , Niels E. Skakkebaek, Richard M. Sharpe JCI Insight. 2017;2(6):e91204. https://doi.org/10.1172/jci.insight.91204. Research Article Endocrinology Reproductive biology The testicular dysgenesis syndrome (TDS) hypothesis, which proposes that common reproductive disorders of newborn and adult human males may have a common fetal origin, is largely untested. We tested this hypothesis using a rat model involving gestational exposure to dibutyl phthalate (DBP), which suppresses testosterone production by the fetal testis. We evaluated if induction of TDS via testosterone suppression is restricted to the “masculinization programming window” (MPW), as indicated by reduction in anogenital distance (AGD). We show that DBP suppresses fetal testosterone equally during and after the MPW, but only DBP exposure in the MPW causes reduced AGD, focal testicular dysgenesis, and TDS disorders (cryptorchidism, hypospadias, reduced adult testis size, and compensated adult Leydig cell failure). Focal testicular dysgenesis, reduced size of adult male reproductive organs, and TDS disorders and their severity were all strongly associated with reduced AGD. We related our findings to human TDS cases by demonstrating similar focal dysgenetic changes in testes of men with preinvasive germ cell neoplasia (GCNIS) and in testes of DBP-MPW animals. If our results are translatable to humans, they suggest that identification of potential causes of human TDS disorders should focus on exposures during a human MPW equivalent, especially if negatively associated with offspring AGD. Find the latest version: https://jci.me/91204/pdf RESEARCH ARTICLE Experimentally induced testicular dysgenesis syndrome originates in the masculinization programming window Sander van den Driesche,1 Karen R. -

Surgical Correction of a Penoscrotal Web:A Report of a Case With
www.symbiosisonline.org Symbiosis www.symbiosisonlinepublishing.com Review Article SOJ Surgery Open Access Surgical Correction of a Penoscrotal Web:A Report of a Case with Literature Review Volkan Sarper Erikci1*, Merve Dilara Öney2, Gökhan Köylüoğlu3 1Attending Pediatric Surgeon, Associate Professor of Pediatric Surgery, Sağlık Bilimleri University, TURKEY 2Trainee in Pediatric Surgery, Sağlık Bilimleri University,Turkey 3Professor of Pediatric Surgery, Chief Department of Pediatric Surgery, Katip Çelebi University, Turkey Received: 7 July, 2017; Accepted: 14 September, 2017; Published: 23 September, 2017 *Corresponding author: Volkan Sarper Erikci, Attending Pediatric Surgeon, Associate Professor of Pediatric Surgery, Sağlık Bilimleri University, Kazim Dirik Mah Mustafa Kemal Cad Hakkibey apt. No:45 D.10 35100 Bornova-İzmir. GSM: +90 542 4372747, Business phone: +90 232 4696969, Fax: +90 232 4330756; E-mail: [email protected] and the medical history did not reveal local infection, urinary Abstract retention or chronic urinary dripping. But the parents were Penoscrotal Webbing (PSW) is a penile and scrotal skin anxious because they felt that their child’s penis was too short. In abnormality that is considered in the spectrum of buried penis. Various surgical techniques have been proposed for PSW with on the ventral aspect of the penis solved the problem (Figure 3,4). different terminologies. Herein we present a 7-year-old boy with PSW Withaddition an uneventful to circumcision, postoperative foreskin period,reconstruction the family -

Meeting Report on the NIDDK/AUA Workshop on Congenital Anomalies of External Genitalia: Challenges and Opportunities for Translational Research
UCLA UCLA Previously Published Works Title Meeting report on the NIDDK/AUA Workshop on Congenital Anomalies of External Genitalia: challenges and opportunities for translational research. Permalink https://escholarship.org/uc/item/1vk2c98g Journal Journal of pediatric urology, 16(6) ISSN 1477-5131 Authors Stadler, H Scott Peters, Craig A Sturm, Renea M et al. Publication Date 2020-12-01 DOI 10.1016/j.jpurol.2020.09.012 Peer reviewed eScholarship.org Powered by the California Digital Library University of California Journal of Pediatric Urology (2020) 16, 791e804 Review Article Meeting report on the NIDDK/AUA Workshop on Congenital Anomalies of External Genitalia: challenges and opportunities for translational research* aDepartment of Skeletal Biology, Shriners Hospital for Children, a, ,1 b,c, ,1 d,1 3101 SW Sam Jackson Park Road, H. Scott Stadler *** , Craig A. Peters ** , Renea M. Sturm , b e f,g Portland, OR, Oregon Health & Linda A. Baker , Carolyn J.M. Best , Victoria Y. Bird , Science University, Department of h i j Orthopaedics and Rehabilitation, Frank Geller , Deborah K. Hoshizaki , Thomas B. Knudsen , i k l, ,1 Portland, 97239, OR, USA Jenna M. Norton , Rodrigo L.P. Romao , Martin J. Cohn * b Department of Urology, Summary parents, and short interviews to determine familial University of Texas Southwestern, 5323 Harry Hines Blvd., Dallas, Congenital anomalies of the external genitalia penetrance (small pedigrees), would accelerate 75390-9110, TX, USA (CAEG) are a prevalent and serious public health research in this field. Such a centralized datahub concern with lifelong impacts on the urinary func- will advance efforts to develop detailed multi- cPediatric Urology, Children’s tion, sexual health, fertility, tumor development, dimensional phenotyping and will enable access to Health System Texas, University of and psychosocial wellbeing of affected individuals. -

Chordee Repair Post-Operative Care
Jose C. Cortez, MD Children’s Urology George M. Seremetis, MD 1301 Barbara Jordan Blvd., Suite 302 Leslie T. McQuiston, MD Austin, Texas 78723 Vani S. Menon, MD PHONE: (512) 472-6134 Kelly J. Nast, MD FAX: (512) 472-2928 Mary “Katie” Wang, MD After Hours: (512) 406-3112 www.childrensurology.com Chordee Repair Post-Operative Care ACTIVITY RESTRICTIONS For the first one or two days following surgery, your child may not feel like being very active. He may increase his level of activity as the soreness goes away. However, for at least 2 weeks (up to 4 weeks depending on post-operative swelling) your son should not be allowed to participate in any activities which would require him to straddle an object, such as a walker, tricycle, or bicycle. If your child is older any strenuous activity, including heavy lifting, contact sports, and gym class should be avoided for the same period of time. No swimming for 2 weeks after the procedure. School age children may return to school after 48 hours. DIET Before your child is discharged home, he should be able to drink clear liquids and keep them down without vomiting. Once your child is able to drink liquids, you may add progressively to his diet with: 1. A full liquid or “light” meal, which may include toast, crackers, soup, or gelatin 2. Regular meals if your child tolerates the above If your child vomits, wait approximately 45 minutes and start this process over with sips of clear liquids. Gradually increase the amount of clear liquids. When your child is able to tolerate them, follow the guidelines above. -

Penile Anomalies in Childhood
Penile Anomalies in Childhood Sarah M. Lambert, MD Assistant Professor of Urology Yale School of Medicine Yale New Haven Health System The Newborn Penis − Development − 9-13 weeks gestation − Testosterone and dihydrotestosterone dependent − Genital tubercle -> glans penis − Genital folds -> penile shaft − Genital swellings -> scrotum − Normal, full-term neonate − Stretched penile length 3.5 cm +/- 0.7 cm − 1.1 cm +/- 0.2 cm diameter The Newborn Penis − Complete foreskin, physiologic phimosis − Median raphe − Deviated 10% − Penile anomalies − Buried penis − Webbed penis − Torsion − Curvature 0.6% male neonates − Hypospadias 1:250 − Epispadias 1:117,000 − Penile anomalies can be associated with anorectal malformations and urologic abnormalities Buried Penis − Abnormal fascial attachments, deficit in penile skin? − CONTRAINDICATION TO NEWBORN CIRCUMCISION − Not MICROPENIS − <2cm stretched penile length − Hypogonadotropic hypogonadism − Testicular failure − Androgen receptor defect − 5 alpha reductase deficit Webbed Penis − Web of skin obscures the penoscrotal junction − Deficit in ventral preputial skin − CONTRAINDICATION TO NEWBORN CIRCUMCISION Penile Torsion and Wandering Raphe • Counterclockwise • Abnormal arrangement of penile shaft skin in development • Surgical repair if >40 degrees Penile curvature − 0.6% incidence − 8.6% penile anomalies − Often associated with hypospadias − Can be initially noted in adolescence with erection − Ventral skin deficiency − Corporeal disproportion Hypospadias − Incomplete virilization of the pubic tubercle -
Postmedj00529-0038.Pdf
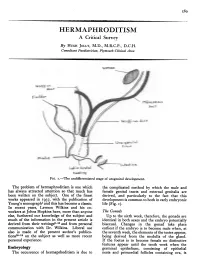
.........M...... I . ' . - HERMAPHRODITISM A Critical Survey By HUGH JOLLY, M.D., M.R.C.P., D.C.H. Consultant Paediatrician, Plymouth Clinical Area Wo-ff ,.,/i/, decttPcradft$ Wrolff LO,^ uct Lsb(oastrhhpv ~ -.I ,~o5: U-e^latiit Iws l FIG. I.-The undifferentiated stage of urogenital development. The problem of hermaphroditism is one which the complicated method by which the male and has always attracted attention so that much has female genital tracts and external genitalia are been written on the subject. One of the finest derived, and particularly to the fact that this works appeared in 1937, with the publication of development is common to both in early embryonic Young's monograph1 and this has become a classic. life (Fig. I). In recent years, Lawson Wilkins and his co- workers at Johns Hopkins have, more than anyone The Gonads else, furthered our knowledge of the subject and Up to the sixth week, therefore, the gonads are much of the information in the present article is identical in both sexes and the embryo potentially derived from their writings2-15 and from personal bisexual. Changes in the gonad take place communication with Dr. Wilkins. Liberal use earliest if the embryo is to become male when, at also is made of the present author's publica- the seventh week, the elements of the testes appear, tions16-18 on the subject as well as more recent being derived from the medulla of the gland. personal experience. If the foetus is to become female no distinctive features appear until the tenth week when the Embryology germinal epithelium, consisting of epithelial The occurrence of hermaprhoditism is due to nests and primordial follicles containing ova, is 590 POSTGRADUATE MEDICAL JOURNAL December 1956 '... -

Intersex Genital Mutilations Human Rights Violations of Children with Variations of Sex Anatomy
v 2.0 Intersex Genital Mutilations Human Rights Violations Of Children With Variations Of Sex Anatomy NGO Report to the 2nd, 3rd and 4th Periodic Report of Switzerland on the Convention on the Rights of the Child (CRC) + Supplement “Background Information on IGMs” Compiled by: Zwischengeschlecht.org (Human Rights NGO) Markus Bauer Daniela Truffer Zwischengeschlecht.org P.O.Box 2122 8031 Zurich info_at_zwischengeschlecht.org http://Zwischengeschlecht.org/ http://StopIGM.org/ Intersex.ch (Peer Support Group) Daniela Truffer kontakt_at_intersex.ch http://intersex.ch/ Verein SI Selbsthilfe Intersexualität (Parents Peer Support Group) Karin Plattner Selbsthilfe Intersexualität P.O.Box 4066 4002 Basel info_at_si-global.ch http://si-global.ch/ March 2014 v2.0: Internal links added, some errors and typos corrected. This NGO Report online: http://intersex.shadowreport.org/public/2014-CRC-Swiss-NGO-Zwischengeschlecht-Intersex-IGM_v2.pdf Front Cover Photo: UPR #14, 20.10.2012 Back Cover Photo: CEDAW #43, 25.01.2009 2 Executive Summary Intersex children are born with variations of sex anatomy, including atypical genetic make- up, atypical sex hormone producing organs, atypical response to sex hormones, atypical geni- tals, atypical secondary sex markers. While intersex children may face several problems, in the “developed world” the most pressing are the ongoing Intersex Genital Mutilations, which present a distinct and unique issue constituting significant human rights violations (A). IGMs include non-consensual, medically unnecessary, irreversible, cosmetic genital sur- geries, and/or other harmful medical treatments that would not be considered for “normal” children, without evidence of benefit for the children concerned, but justified by societal and cultural norms and beliefs.