Asprosin Is a Centrally-Acting Orexigenic Hormone
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

03 November 2019 Palm Wings Ephesus Hotel Congress Center, Kus¸Adası, Aydın (Turkey)
www.actaphysiol.org December 2019 • Volume 227 • Supplement 722 OFFICIAL JOURNAL OF THE FEDERATION OF EUROPEAN PHYSIOLOGICAL SOCIETIES Turkish Society of Physiological Sciences 45th National Physiology Congress 31 October – 03 November 2019 Palm Wings Ephesus Hotel Congress Center, Kus¸adası, Aydın (Turkey) PUBLICATION HISTORY Acta Physiologica 2006– Acta Physiologica Scandinavica 1940–2005 Skandinavisches Archiv für Physiologie 1889–1939 AAPHA_v227_is722_issue-info.inddPHA_v227_is722_issue-info.indd 1 229-Nov-199-Nov-19 12:00:2112:00:21 PMPM Chief Editor INFORMATION FOR SUBSCRIBERS Pontus B. Persson, Berlin Acta Physiologica is published in 12 issues per year. Subscription prices for 2020 € Editors are: Institutional: 1112 (Europe), $1662 (The Americas), $1942 (Rest of World). Cardiovascular Physiology – Frantisek Kolar, Prague; Holger Nilsson, Gothenburg Prices are exclusive of tax. Australian GST, Canadian GST/HST and European and William E. Louch, Oslo VAT will be applied at the appropriate rates. For more information on current tax Cell Biology – Sari Lauri, Helsinki rates, please go to www.wileyonlinelibrary.com/tax-vat. The price includes online Chronobiology and Endocrinology – Charna Dibner, Geneva access to the current and all online back fi les to January 1st 2016, where available. Exercise Physiology – Jan Henriksson, Stockholm For other pricing options, including access information and terms and conditions, Gastrointestinal Physiology – Markus Sjöblom, Uppsala please visit www.wileyonlinelibrary.com/access Infl ammation – Joakim Ek, Gothenburg Metabolism and Nutritional Physiology – Karl-Heinz Herzig, Oulu DELIVERY TERMS AND LEGAL TITLE Muscle Physiology – Gabriele Pfi tzer, Cologne Where the subscription price includes print issues and delivery is to the recipients Nervous System – Alexej Verkhratsky, Manchester address, delivery terms are Delivered at Place (DAP); the recipient is responsible Renal Physiology – Peter Bie, Odense; Boye L. -

W W W .Bio Visio N .Co M New Products Added in 2020
New products added in 2020 Please find below a list of all the products added to our portfolio in the year 2020. Assay Kits Product Name Cat. No. Size Product Name Cat. No. Size N-Acetylcysteine Assay Kit (F) K2044 100 assays Human GAPDH Activity Assay Kit II K2047 100 assays Adeno-Associated Virus qPCR Quantification Kit K1473 100 Rxns Human GAPDH Inhibitor Screening Kit (C) K2043 100 assays 20 Preps, Adenovirus Purification Kit K1459 Hydroxyurea Colorimetric Assay Kit K2046 100 assays 100 Preps Iodide Colorimetric Assay Kit K2037 100 assays Aldehyde Dehydrogenase 2 Inhibitor Screening Kit (F) K2011 100 assays Laccase Activity Assay Kit (C) K2038 100 assays Aldehyde Dehydrogenase 3A1 Inhibitor Screening Kit (F) K2060 100 assays 20 Preps, Lentivirus and Retrovirus Purification Kit K1458 Alkaline Phosphatase Staining Kit K2035 50 assays 100 Preps Alpha-Mannosidase Activity Assay Kit (F) K2041 100 assays Instant Lentivirus Detection Card K1470 10 tests, 20 tests Beta-Mannosidase Activity Assay Kit (F) K2045 100 assays Lentivirus qPCR Quantification Kit K1471 100 Rxns 50 Preps, Buccal Swab DNA Purification Kit K1466 Maleimide Activated KLH-Peptide Conjugation Kit K2039 5 columns 250 Preps Methionine Adenosyltransferase Activity Assay Kit (C) K2033 100 assays CD38 Activity Assay Kit (F) K2042 100 assays miRNA Extraction Kit K1456 50 Preps EZCell™ CFDA SE Cell Tracer Kit K2057 200 assays MMP-13 Inhibitor Screening Kit (F) K2067 100 assays Choline Oxidase Activity Assay Kit (F) K2052 100 assays Mycoplasma PCR Detection Kit K1476 100 Rxns Coronavirus -

Follicle-Stimulating Hormone (FSH)
Hormone Structure (1) Principal Source Link to diagram showing locations of the main endocrine glands Thyroid-stimulating hormone (TSH) protein (201) Follicle-stimulating hormone (FSH) protein (204) Luteinizing hormone (LH) protein (204) Anterior lobe of pituitary Prolactin (PRL) protein (198) Growth hormone (GH) protein (191) Adrenocorticotropic hormone (ACTH) peptide (39) Vasopressin peptide (9) Posterior lobe of pituitary Oxytocin peptide (9) Thyrotropin-releasing hormone (TRH) peptide (3) Gonadotropin-releasing hormone (GnRH) peptide (10) Growth hormone-releasing hormone (GHRH) peptides (40, 44) Hypothalamus Corticotropin-releasing hormone (CRH) peptide (41) Somatostatin peptides (14, 28) Dopamine tyrosine derivative Melatonin tryptophan derivative Pineal gland Thyroxine (T4) tyrosine derivative Thyroid Gland Calcitonin peptide (32) Parathyroid hormone (PTH) protein (84) Parathyroid glands FGF-23 (phosphatonin) protein (251) Osteocalcin peptide (49) Bone Lipocalin 2 protein (198) Erythropoietin (EPO) protein (166) Glucocorticoids (e.g., cortisol) steroids Mineralocorticoids (e.g., aldosterone) steroids Adrenal cortex Androgens (e.g., testosterone) steroids Adrenaline (epinephrine) tyrosine derivative Adrenal medulla Noradrenaline (norepinephrine) tyrosine derivative Estrogens (e.g., estradiol) steroid Ovarian follicle Progesterone steroid Corpus luteum and placenta Human chorionic gonadotropin (HCG) protein (237) Trophoblast and placenta Androgens (e.g., testosterone) steroid Testes Insulin protein (51) Glucagon peptide (29) Pancreas -

The Impaired Response of Circulating Asprosin Concentrations to Glucose Levels Fluctuation May Be One of the Causes of Type 2 Diabetes – a Narrative Review
Trojanowska Paulina, Trojanowska Alina, Szydełko Joanna, Tywanek Ewa, Łuczyk Robert Jan. The impaired response of circulating asprosin concentrations to glucose levels fluctuation may be one of the causes of type 2 diabetes – a narrative review. Journal of Education, Health and Sport. 2020;10(9):846-854. eISSN 2391-8306. DOI http://dx.doi.org/10.12775/JEHS.2020.10.09.10 2 https://apcz.umk.pl/czasopisma/index.php/JEHS/article/view/JEHS.2020.10.09. 10 2 https://zenodo.org/record/4054287 The journal has had 5 points in Ministry of Science and Higher Education parametric evaluation. § 8. 2) and § 12. 1. 2) 22.02.2019. © The Authors 2020; This article is published with open access at Licensee Open Journal Systems of Nicolaus Copernicus University in Torun, Poland Open Access. This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author (s) and source are credited. This is an open access article licensed under the terms of the Creative Commons Attribution Non commercial license Share alike. (http://creativecommons.org/licenses/by-nc-sa/4.0/) which permits unrestricted, non commercial use, distribution and reproduction in any medium, provided the work is properly cited. The authors declare that there is no conflict of interests regarding the publication of this paper. Received: 21.09.2020. Revised: 27.09.2020. Accepted: 28.09.2020. The impaired response of circulating asprosin concentrations -

Asprosin-Neutralizing Antibodies As a Treatment for Metabolic Syndrome
RESEARCH ARTICLE Asprosin-neutralizing antibodies as a treatment for metabolic syndrome Ila Mishra1†, Clemens Duerrschmid1†, Zhiqiang Ku2, Yang He3, Wei Xie1, Elizabeth Sabath Silva1, Jennifer Hoffman1, Wei Xin4, Ningyan Zhang2, Yong Xu3, Zhiqiang An2, Atul R Chopra1,5,6* 1Harrington Discovery Institute, University Hospitals, Cleveland, United States; 2Texas Therapeutics Institute, Brown Foundation Institute of Molecular Medicine, University of Texas Health Science Center at Houston, Houston, United States; 3Baylor College of Medicine, Houston, United States; 4Department of Pathology, Case Western Reserve University, Cleveland, United States; 5Department of Medicine, Case Western Reserve University, Cleveland, United States; 6Department of Genetics and Genome Sciences, Case Western Reserve University, Cleveland, United States Abstract Background: Recently, we discovered a new glucogenic and centrally acting orexigenic hormone – asprosin. Asprosin is elevated in metabolic syndrome (MS) patients, and its genetic loss results in reduced appetite, leanness, and blood glucose burden, leading to protection from MS. Methods: We generated three independent monoclonal antibodies (mAbs) that recognize unique asprosin epitopes and investigated their preclinical efficacy and tolerability in the treatment of MS. Results: Anti-asprosin mAbs from three distinct species lowered appetite and body weight, and reduced blood glucose in a dose-dependent and epitope-agnostic fashion in three independent MS mouse models, with an IC50 of ~1.5 mg/kg. The mAbs displayed a half-life of over 3days in vivo, *For correspondence: with equilibrium dissociation-constants in picomolar to low nanomolar range. [email protected] Conclusions: We demonstrate that anti-asprosin mAbs are dual-effect pharmacologic therapy that †These authors contributed targets two key pillars of MS – over-nutrition and hyperglycemia. -

Effect of Swimming Training on Levels of Asprosin, Lipid Profile, Glucose and Insulin Resistance in Rats with Metabolic Syndrome
Obesity Medicine 15 (2019) 100111 Contents lists available at ScienceDirect Obesity Medicine journal homepage: www.elsevier.com/locate/obmed Original research Effect of swimming training on levels of asprosin, lipid profile, glucose and insulin resistance in rats with metabolic syndrome T ∗ Hossein Nakhaeia, Mehdi Mogharnasia, Hamed Fanaeib,c, a Department of Sport Sciences, Faculty of Sport Sciences, University of Birjand, Birjand, Iran b Pregnancy Health Research Center, Zahedan University of Medical Sciences, Zahedan, Iran c Department of Physiology, School of Medicine, Zahedan University of Medical Sciences, Zahedan, Iran ARTICLE INFO ABSTRACT Keywords: Background: Asprosin is a novel biomarker that associated with type 2 diabetes mellitus. The aim of this study Continuous training was to investigate the effects of continuous and interval swimming training on the asprosin, lipid profile, glucose Interval training concentration and insulin resistance serum levels of rats with metabolic syndrome. Asprosin Methods: Forty-eight male Wistar rats were randomly divided into two groups, standard diet (SD) and high-fat Lipid profile diet (HD), and received their respective diets for a period of 12 weeks without exercise stimuli. After this period, Metabolic syndrome the animals were randomly divided into four groups (n = 8); normal control standard diet (NC), control (Ctr), continuous training (load 0–3% body mass, 5 d/wk, for 8 weeks, CT) and interval training (load 5–16% body mass, 5 d/wk, for 8 weeks, IT). The continuous and interval training consisted of a swimming exercise performed over eight weeks. Result: The NC and trained groups showed lower values of total cholesterol (TC), triglyceride (TG), low-density lipoprotein (LDL-C) and glucose concentration compared to Ctr group. -

Serum Concentration of Asprosin in New-Onset Type 2 Diabetes
Naiemian et al. Diabetol Metab Syndr (2020) 12:65 https://doi.org/10.1186/s13098-020-00564-w Diabetology & Metabolic Syndrome RESEARCH Open Access Serum concentration of asprosin in new-onset type 2 diabetes Shakiba Naiemian1, Mohsen Naeemipour2, Mehdi Zarei3, Moslem Lari Najaf4, Ali Gohari2, Mohammad Reza Behroozikhah2, Hafez Heydari2* and Mohammad Miri5* Abstract Background: Asprosin, a newly identifed adipokine, is pathologically increased in individuals with insulin resist- ance. However, the available evidence on the association of asprosin and type 2 diabetes mellitus (T2DM) status is still scarce. Therefore, this study aimed to determine the relationship between serum concentrations of asprosin and T2DM status. Methods: This observational study was performed based on 194 adults (97 newly diagnosed T2DM and 97 healthy individuals). Anthropometric and biochemical variables were determined in all participants. Serum concentrations of asprosin were measured using enzyme-linked immunosorbent assay (ELISA). Results: In patients with T2DM, the serum concentrations of asprosin were signifcantly higher than the healthy con- trols (4.18 [IQR: 4.4] vs. 3.5 [IQR: 1.85], P < 0.001). The concentrations of asprosin were signifcantly correlated with body mass index (BMI) and fasting blood glucose (FBG) in healthy subjects and with BMI, FBG, hemoglobin A1c (HbA1c), homeostatic model assessment of insulin resistance (HOMA-IR), and quantitative insulin check index (QUICKI), triacyl- glycerol (TAG) and total cholesterol/high-density lipoprotein cholesterol (TC/HDL-C) ratio in the T2DM group. In fully adjusted model, the odds ratio (OR) of T2DM with serum concentrations of asprosin was approximately 1.547 (95% CI 1.293–1.850, P < 0.001) compared to the control group. -

An Investigation of the Relationship Between New Fasting Hormone Asprosin, Obesity and Acute–Chronic Exercise: Current Systematic Review
ARCHIVES OF PHYSIOLOGY AND BIOCHEMISTRY https://doi.org/10.1080/13813455.2020.1767652 REVIEW ARTICLE An investigation of the relationship between new fasting hormone asprosin, obesity and acute–chronic exercise: current systematic review Halil _Ibrahim Ceylana and Ozcan€ Saygınb aFaculty of Kazim Karabekir Education, Physical Education and Sports Teaching Department, Ataturk University, Erzurum, Turkey; bFaculty of Sports Sciences, Coaching Science Department, Mugla Sitki Kocman University, Mugla, Turkey ABSTRACT ARTICLE HISTORY The purpose of this study was to reveal the relationship between new fasting hormone asprosin, obes- Received 31 March 2020 ity, and acute–chronic exercise. The prisma guidelines were followed in forming the methodological Revised 5 May 2020 model of this review. The articles between 2016 and 2020 (including March) were identified by scan- Accepted 7 May 2020 ning Google Scholar, Pub Med, and Science Direct databases. Thirty-five articles were defined from Published online 18 May 188 articles. Three cross-sectional, and 1 prospective cohort design studies in adults, and 3 cross-sec- 2020 tional studies in children were found. Three randomised-control group designed studies which exam- KEYWORDS ined the effect of acute exercise on serum asprosin levels in obese individuals. Asprosin may be a new Appetite; asprosin; exercise; therapeutic biomarker to be considered in the development, but long-term and deep-rooted obesity; systematic review researches are needed, and increasing the number of studies examining the effect of exercise on asprosin in the future might help us to identify the mechanisms underlying the decrease or increase in asprosin after exercise. Introduction obesity, it is necessary to understand especially how appe- tite/nutritional behaviours are regulated in the control of Obesity is a global public health problem, and ranked 5th body weight, and this also helps to develop new strategies with 4.8% of deaths worldwide (Wilson et al. -
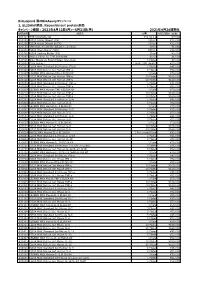
Recombinant Protein製品 キャンペーン期間
BioLegend 春のBioAssayキャンペーン 1, ELISAkit製品、Recombinant protein製品 キャンペーン期間:2021年4月12日(月)~5月31日(月) 2021年4月26日現在 製品番号 製品名 容量 カタログ価格(税抜) 421101 TMB Substrate Set 110 ml x 2 ¥7,000 421203 ELISA Assay Diluent (5X) 100 ml ¥12,000 421205 ELISA Assay Diluent B (5X) 100 ml ¥12,000 421501 TMB High Sensitivity Substrate Solution 60 ml ¥5,000 421601 ELISA Wash Buffer (20X) 500 ml ¥24,000 421701 ELISA Coating Buffer (5X) 30 ml ¥3,000 423001 Stop Solution for TMB Substrate 60 ml ¥6,000 423501 Nunc MaxiSorp ELISA Plates, Uncoated 5 Plates ¥7,000 423601 Plate Sealers 1 pack (100 sheets) ¥13,000 430101 ELISA MAX Standard Set Human IFN-g 5 Plates ¥45,000 430104 ELISA MAX Deluxe Set Human IFN-g 5 Plates ¥52,000 430107 LEGEND MAX Human IFN-g ELISA Kit 1 Plate ¥70,000 430115 ELISA MAX Deluxe Set Human IFN-g 10 Plates ¥100,000 430116 ELISA MAX Deluxe Set Human IFN-g 20 Plates ¥160,000 430201 ELISA MAX Standard Set Human TNF-a 5 Plates ¥45,000 430204 ELISA MAX Deluxe Set Human TNF-a 5 Plates ¥52,000 430207 LEGEND MAX Human TNF-a ELISA Kit 1 Plate ¥70,000 430215 ELISA MAX Deluxe Set Human TNF-a 10 Plates ¥100,000 430216 ELISA MAX Deluxe Set Human TNF-a 20 Plates ¥160,000 430301 ELISA MAX Standard Set Human IL-4 5 Plates ¥45,000 430304 ELISA MAX Deluxe Set Human IL-4 5 Plates ¥52,000 430307 LEGEND MAX Human IL-4 ELISA Kit 1 Plate ¥70,000 430401 ELISA MAX Standard Set Human IL-5 5 Plates ¥45,000 430404 ELISA MAX Deluxe Set Human IL-5 5 Plates ¥52,000 430501 ELISA MAX Standard Set Human IL-6 5 Plates ¥45,000 430504 ELISA MAX Deluxe Set Human IL-6 5 Plates ¥52,000 430507 -

Correlation Between Serum Asprosin Level and Oxidative Stress in Iraqi
Sys Rev Pharm 2020;11(12):1729-1733 A multifaceted review journal in the field of pharmacy Correlation Between Serum Asprosin Level And Oxidative Stress In Iraqi Patients With Type Ii Diabetes Mellitus Mohammed Basim Ahmed Alobaidi and Rafah Razooq Hameed Al-Samarrai Department of chemistry, College of Education, University of Samarra, Salah Eldin, Iraq. Corresponding author E-mail: [email protected] Abstract Background: Asprosin, a new protein hormone identifies by Keywords: asprosin, total antioxidant capacity, diabetic USA researcher team in 2016, secreted by the white adipose mellitus, Glutathione-S-transferase. tissue. Asprosin induce releasing of hepatic glucose, its pathologically increased in insulin resistant, obesity and diabetic mellitus. Method: Cross sectional study were carried out to evaluate the concentration of serum asprosin, glucose, Total antioxidant capacity-TAC, malondialdehyde –MDA, Glutathione-S- transferase-GST and HbA1c for 121 sample of patients with type II diabetes mellitus (age between 25-45) collected from Specialized Center for Diabetes and Endocrinology / Baghdad- Alrasapha from 1/10/2019 to 20/12/2019. The groups of samples were divided into three groups, 1st group G1 include twenty four patients(12male and 12female) newly diagnose with type II diabetic mellitus without any treatment(new onset), 2nd group G2 include sixty nine(36male and 33female) patients with type II diabetic mellitus taken a treatment(old diagnostic cases) and twenty eight sample(12male and 16 female) for healthy individuals as control group-C. Results:The results showed that the concentration of asprosin, glucose, HbA1c and MDA were significantly higher P≤0.05 in G2 as compared with control group, with no significant change P≤0.05 to the concentration of asprosin , glucose and MDA in G1 as compared with control group, while at the same group the concentration of HbA1c and GST significantly higher P≤0.05 comparing with C, The concentration of TAC significantly decreased P≤0.05 in G1 and G2 as comparing with control group. -

Circulating Levels of Asprosin and Its Association with Insulin Resistance and Renal Function in Type2 Diabetes Mellitus and Diabetic Nephropathy Patients
Circulating levels of asprosin and its association with insulin resistance and renal function in type2 diabetes mellitus and diabetic nephropathy patients Golnoosh Goodarzi Islamic Azad University Leila Setayesh Tehran University of Medical Sciences Reza Fadaei Kermanshah University of Medical Sciences Mohammad Ebrahim Khamseh Iran University of Medical Sciences Fereshteh Aliakbari Shahid Beheshti University of Medical Sciences School of Traditional Medicine Jalil Hosseini Shahid Beheshti University of Medical Sciences School of Medicine Nariman Moradi ( [email protected] ) Kurdistan University of Medical Sciences https://orcid.org/0000-0001-7760-4804 Research Article Keywords: Asprosin, Type 2 diabetes, Diabetic nephropathy, Adipokine, Insulin resistance Posted Date: May 5th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-448826/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Version of Record: A version of this preprint was published at Molecular Biology Reports on July 25th, 2021. See the published version at https://doi.org/10.1007/s11033-021-06551-2. Page 1/13 Abstract Introduction : Adipokines have an important role in development and progression of type 2 diabetes mellitus (T2DM) and its complications such as nephropathy. Asprosin is a recently discovered adipokine that involve in glucose metabolism and inammation process. The present study sought to evaluate asprosin levels in patients with T2DM and T2DM + nephropathy (NP) compared to controls and its relation with markers of insulin resistance, inammation and renal function. Methods Serum levels of asprosin, adiponectin, IL-6 and TNF-α were measured in 55 control, 54 T2DM and 55 T2DM + NP patients using ELISA kits. -

Asprosin Dynamics Relating to Serum Glucose Levels Under Controlled Manipulation
Asprosin dynamics relating to serum Glucose levels under controlled alteration Study protocol version 1.4 Heidelberg, 31/05/2017 Asprosin dynamics relating to serum Glucose levels under controlled alteration from Version 1.1 Stand 31.05.17 1 Table of contents Page 1 Title of the study 3 2 Principal investigator and coordination 3 2.1 Principal investigator 3 2.2 Study coordination and study doctors 3 3 Summary 4 4 Scientific background and state of the art 6 5 Aims and hypotheses of the study 9 6 Study design and examinations 10 6.1 Study examinations 11 Module 1: Medical history (questionnaire) and clinical basic testing (mandatory) 11 Module 2: Blood and urine testing (mandatory) 11 Module 3: insulin resistance module and Asprosin kinetics 12 Module 4: neuropathy module (optional) 14 Module 5: Vascular module (mandatory) 20 Module 6: Pulmonary examination 21 6.2 Duration of the study 21 6.3 Selection of study participants 21 6.4 Course of the study 23 6.5 Potential complications and risks 23 6.6 Fallout Criteria 24 6.7 Concomitant therapy 24 6.8 Sponsor 24 7 Statistical analysis and sample size calculation 24 8 Data collection and data management 25 9 Legal regulations and insurance 26 Asprosin dynamics relating to serum Glucose levels under controlled alteration from Version 1.1 Stand 31.05.17 2 Title of the study Asprosin dynamics relating to serum Glucose levels under controlled alteration 2. Principal investigator and coordination 2.1 Principal investigator: Dr. Stefan Kopf Attending physician Internal Medicine I and clinical chemistry Medical faculty of the University hospital Heidelberg Im Neuenheimer Feld 410 69120 Heidelberg Phone: 06221 – 56 – 37790, fax 06221 – 56 – 4233 [email protected] Signature principal investigator: __________________________ 2.2 Study coordination and study doctors Valkanou Aikaterini (study doctor) Internal Medicine I and clinical chemistry Medical faculty of the University hospital Heidelberg Im Neuenheimer Feld 410 69120 Heidelberg Phone: 06221 – 56 –37940, fax 06221 – 56 – 4233 Dr.