Probability and Statistics for Bioinformatics and Genetics Course
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bioinformatics 1
Bioinformatics 1 Bioinformatics School School of Science, Engineering and Technology (http://www.stmarytx.edu/set/) School Dean Ian P. Martines, Ph.D. ([email protected]) Department Biological Science (https://www.stmarytx.edu/academics/set/undergraduate/biological-sciences/) Bioinformatics is an interdisciplinary and growing field in science for solving biological, biomedical and biochemical problems with the help of computer science, mathematics and information technology. Bioinformaticians are in high demand not only in research, but also in academia because few people have the education and skills to fill available positions. The Bioinformatics program at St. Mary’s University prepares students for graduate school, medical school or entry into the field. Bioinformatics is highly applicable to all branches of life sciences and also to fields like personalized medicine and pharmacogenomics — the study of how genes affect a person’s response to drugs. The Bachelor of Science in Bioinformatics offers three tracks that students can choose. • Bachelor of Science in Bioinformatics with a minor in Biology: 120 credit hours • Bachelor of Science in Bioinformatics with a minor in Computer Science: 120 credit hours • Bachelor of Science in Bioinformatics with a minor in Applied Mathematics: 120 credit hours Students will take 23 credit hours of core Bioinformatics classes, which included three credit hours of internship or research and three credit hours of a Bioinformatics Capstone course. BS Bioinformatics Tracks • Bachelor of Science -

13 Genomics and Bioinformatics
Enderle / Introduction to Biomedical Engineering 2nd ed. Final Proof 5.2.2005 11:58am page 799 13 GENOMICS AND BIOINFORMATICS Spencer Muse, PhD Chapter Contents 13.1 Introduction 13.1.1 The Central Dogma: DNA to RNA to Protein 13.2 Core Laboratory Technologies 13.2.1 Gene Sequencing 13.2.2 Whole Genome Sequencing 13.2.3 Gene Expression 13.2.4 Polymorphisms 13.3 Core Bioinformatics Technologies 13.3.1 Genomics Databases 13.3.2 Sequence Alignment 13.3.3 Database Searching 13.3.4 Hidden Markov Models 13.3.5 Gene Prediction 13.3.6 Functional Annotation 13.3.7 Identifying Differentially Expressed Genes 13.3.8 Clustering Genes with Shared Expression Patterns 13.4 Conclusion Exercises Suggested Reading At the conclusion of this chapter, the reader will be able to: & Discuss the basic principles of molecular biology regarding genome science. & Describe the major types of data involved in genome projects, including technologies for collecting them. 799 Enderle / Introduction to Biomedical Engineering 2nd ed. Final Proof 5.2.2005 11:58am page 800 800 CHAPTER 13 GENOMICS AND BIOINFORMATICS & Describe practical applications and uses of genomic data. & Understand the major topics in the field of bioinformatics and DNA sequence analysis. & Use key bioinformatics databases and web resources. 13.1 INTRODUCTION In April 2003, sequencing of all three billion nucleotides in the human genome was declared complete. This landmark of modern science brought with it high hopes for the understanding and treatment of human genetic disorders. There is plenty of evidence to suggest that the hopes will become reality—1631 human genetic diseases are now associated with known DNA sequences, compared to the less than 100 that were known at the initiation of the Human Genome Project (HGP) in 1990. -

Use of Bioinformatics Resources and Tools by Users of Bioinformatics Centers in India Meera Yadav University of Delhi, [email protected]
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Library Philosophy and Practice (e-journal) Libraries at University of Nebraska-Lincoln 2015 Use of Bioinformatics Resources and Tools by Users of Bioinformatics Centers in India meera yadav University of Delhi, [email protected] Manlunching Tawmbing Saha Institute of Nuclear Physisc, [email protected] Follow this and additional works at: http://digitalcommons.unl.edu/libphilprac Part of the Library and Information Science Commons yadav, meera and Tawmbing, Manlunching, "Use of Bioinformatics Resources and Tools by Users of Bioinformatics Centers in India" (2015). Library Philosophy and Practice (e-journal). 1254. http://digitalcommons.unl.edu/libphilprac/1254 Use of Bioinformatics Resources and Tools by Users of Bioinformatics Centers in India Dr Meera, Manlunching Department of Library and Information Science, University of Delhi, India [email protected], [email protected] Abstract Information plays a vital role in Bioinformatics to achieve the existing Bioinformatics information technologies. Librarians have to identify the information needs, uses and problems faced to meet the needs and requirements of the Bioinformatics users. The paper analyses the response of 315 Bioinformatics users of 15 Bioinformatics centers in India. The papers analyze the data with respect to use of different Bioinformatics databases and tools used by scholars and scientists, areas of major research in Bioinformatics, Major research project, thrust areas of research and use of different resources by the user. The study identifies the various Bioinformatics services and resources used by the Bioinformatics researchers. Keywords: Informaion services, Users, Inforamtion needs, Bioinformatics resources 1. Introduction ‘Needs’ refer to lack of self-sufficiency and also represent gaps in the present knowledge of the users. -

Challenges in Bioinformatics
Yuri Quintana, PhD, delivered a webinar in AllerGen’s Webinars for Research Success series on February 27, 2018, discussing different approaches to biomedical informatics and innovations in big-data platforms for biomedical research. His main messages and a hyperlinked index to his presentation follow. WHAT IS BIOINFORMATICS? Bioinformatics is an interdisciplinary field that develops analytical methods and software tools for understanding clinical and biological data. It combines elements from many fields, including basic sciences, biology, computer science, mathematics and engineering, among others. WHY DO WE NEED BIOINFORMATICS? Chronic diseases are rapidly expanding all over the world, and associated healthcare costs are increasing at an astronomical rate. We need to develop personalized treatments tailored to the genetics of increasingly diverse patient populations, to clinical and family histories, and to environmental factors. This requires collecting vast amounts of data, integrating it, and making it accessible and usable. CHALLENGES IN BIOINFORMATICS Data collection, coordination and archiving: These technologies have evolved to the point Many sources of biomedical data—hospitals, that we can now analyze not only at the DNA research centres and universities—do not have level, but down to the level of proteins. DNA complete data for any particular disease, due in sequencers, DNA microarrays, and mass part to patient numbers, but also to the difficulties spectrometers are generating tremendous of data collection, inter-operability -

Regulatory Motifs of DNA
Lecture 3: PWMs and other models for regulatory elements in DNA -Sequence motifs -Position weight matrices (PWMs) -Learning PWMs combinatorially, or probabilistically -Learning from an alignment -Ab initio learning: Baum-Welch & Gibbs sampling -Visualization of PWMs as sequence logos -Search methods for PWM occurrences -Cis-regulatory modules Regulatory motifs of DNA Measuring gene activity (’expression’) with a microarray 1 Jointly regulated gene sets Cluster of co-expressed genes, apply pattern discovery in regulatory regions 600 basepairs Expression profiles Retrieve Upstream regions Genes with similar expression pattern Pattern over-represented in cluster Find a hidden motif 2 Find a hidden motif Find a hidden motif (cont.) Multiple local alignment! Smith-Waterman?? 3 Motif? • Definition: Motif is a pattern that occurs (unexpectedly) often in a string or in a set of strings • The problem of finding repetitions is similar cgccgagtgacagagacgctaatcagg ctgtgttctcaggatgcgtaccgagtg ggagacagcagcacgaccagcggtggc agagacccttgcagacatcaagctctt tgggaacaagtggagcaccgatgatgt acagccgatcaatgacatttccctaat gcaggattacattgcagtgcccaagga gaagtatgccaagtaatacctccctca cagtg... 4 Longest repeat? Example: A 50 million bases long fragment of human genome. We found (using a suffix-tree) the following repetitive sequence of length 2559, with one occurrence at 28395980, and other at 28401554r ttagggtacatgtgcacaacgtgcaggtttgttacatatgtatacacgtgccatgatggtgtgctgcacccattaactcgtcatttagcgttaggtatatctcc gaatgctatccctcccccctccccccaccccacaacagtccccggtgtgtgatgttccccttcctgtgtccatgtgttctcattgttcaattcccacctatgagt -

Comparing Bioinformatics Software Development by Computer Scientists and Biologists: an Exploratory Study
Comparing Bioinformatics Software Development by Computer Scientists and Biologists: An Exploratory Study Parmit K. Chilana Carole L. Palmer Amy J. Ko The Information School Graduate School of Library The Information School DUB Group and Information Science DUB Group University of Washington University of Illinois at University of Washington [email protected] Urbana-Champaign [email protected] [email protected] Abstract Although research in bioinformatics has soared in the last decade, much of the focus has been on high- We present the results of a study designed to better performance computing, such as optimizing algorithms understand information-seeking activities in and large-scale data storage techniques. Only recently bioinformatics software development by computer have studies on end-user programming [5, 8] and scientists and biologists. We collected data through semi- information activities in bioinformatics [1, 6] started to structured interviews with eight participants from four emerge. Despite these efforts, there still is a gap in our different bioinformatics labs in North America. The understanding of problems in bioinformatics software research focus within these labs ranged from development, how domain knowledge among MBB computational biology to applied molecular biology and and CS experts is exchanged, and how the software biochemistry. The findings indicate that colleagues play a development process in this domain can be improved. significant role in information seeking activities, but there In our exploratory study, we used an information is need for better methods of capturing and retaining use perspective to begin understanding issues in information from them during development. Also, in bioinformatics software development. We conducted terms of online information sources, there is need for in-depth interviews with 8 participants working in 4 more centralization, improved access and organization of bioinformatics labs in North America. -

Database Technology for Bioinformatics from Information Retrieval to Knowledge Systems
Database Technology for Bioinformatics From Information Retrieval to Knowledge Systems Luis M. Rocha Complex Systems Modeling CCS3 - Modeling, Algorithms, and Informatics Los Alamos National Laboratory, MS B256 Los Alamos, NM 87545 [email protected] or [email protected] 1 Molecular Biology Databases 3 Bibliographic databases On-line journals and bibliographic citations – MEDLINE (1971, www.nlm.nih.gov) 3 Factual databases Repositories of Experimental data associated with published articles and that can be used for computerized analysis – Nucleic acid sequences: GenBank (1982, www.ncbi.nlm.nih.gov), EMBL (1982, www.ebi.ac.uk), DDBJ (1984, www.ddbj.nig.ac.jp) – Amino acid sequences: PIR (1968, www-nbrf.georgetown.edu), PRF (1979, www.prf.op.jp), SWISS-PROT (1986, www.expasy.ch) – 3D molecular structure: PDB (1971, www.rcsb.org), CSD (1965, www.ccdc.cam.ac.uk) Lack standardization of data contents 3 Knowledge Bases Intended for automatic inference rather than simple retrieval – Motif libraries: PROSITE (1988, www.expasy.ch/sprot/prosite.html) – Molecular Classifications: SCOP (1994, www.mrc-lmb.cam.ac.uk) – Biochemical Pathways: KEGG (1995, www.genome.ad.jp/kegg) Difference between knowledge and data (semiosis and syntax)?? 2 Growth of sequence and 3D Structure databases Number of Entries 3 Database Technology and Bioinformatics 3 Databases Computerized collection of data for Information Retrieval Shared by many users Stored records are organized with a predefined set of data items (attributes) Managed by a computer program: the database -

Motif Discovery in Sequential Data Kyle L. Jensen
Motif discovery in sequential data by Kyle L. Jensen Submitted to the Department of Chemical Engineering in partial fulfillment of the requirements for the degree of Doctor of Philosophy at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY May © Kyle L. Jensen, . Author.............................................................. Department of Chemical Engineering April , Certified by . Gregory N. Stephanopoulos Bayer Professor of Chemical Engineering esis Supervisor Accepted by . William M. Deen Chairman, Department Committee on Graduate Students Motif discovery in sequential data by Kyle L. Jensen Submitted to the Department of Chemical Engineering on April , , in partial fulfillment of the requirements for the degree of Doctor of Philosophy Abstract In this thesis, I discuss the application and development of methods for the automated discovery of motifs in sequential data. ese data include DNA sequences, protein sequences, and real– valued sequential data such as protein structures and timeseries of arbitrary dimension. As more genomes are sequenced and annotated, the need for automated, computational methods for analyzing biological data is increasing rapidly. In broad terms, the goal of this thesis is to treat sequential data sets as unknown languages and to develop tools for interpreting an understanding these languages. e first chapter of this thesis is an introduction to the fundamentals of motif discovery, which establishes a common mode of thought and vocabulary for the subsequent chapters. One of the central themes of this work is the use of grammatical models, which are more commonly associated with the field of computational linguistics. In the second chapter, I use grammatical models to design novel antimicrobial peptides (AmPs). AmPs are small proteins used by the innate immune system to combat bacterial infection in multicellular eukaryotes. -

A General Pairwise Interaction Model Provides an Accurate Description of in Vivo Transcription Factor Binding Sites
A General Pairwise Interaction Model Provides an Accurate Description of In Vivo Transcription Factor Binding Sites Marc Santolini, Thierry Mora, Vincent Hakim* Laboratoire de Physique Statistique, CNRS, Universite´ P. et M. Curie, Universite´ D. Diderot, E´cole Normale Supe´rieure, Paris, France Abstract The identification of transcription factor binding sites (TFBSs) on genomic DNA is of crucial importance for understanding and predicting regulatory elements in gene networks. TFBS motifs are commonly described by Position Weight Matrices (PWMs), in which each DNA base pair contributes independently to the transcription factor (TF) binding. However, this description ignores correlations between nucleotides at different positions, and is generally inaccurate: analysing fly and mouse in vivo ChIPseq data, we show that in most cases the PWM model fails to reproduce the observed statistics of TFBSs. To overcome this issue, we introduce the pairwise interaction model (PIM), a generalization of the PWM model. The model is based on the principle of maximum entropy and explicitly describes pairwise correlations between nucleotides at different positions, while being otherwise as unconstrained as possible. It is mathematically equivalent to considering a TF-DNA binding energy that depends additively on each nucleotide identity at all positions in the TFBS, like the PWM model, but also additively on pairs of nucleotides. We find that the PIM significantly improves over the PWM model, and even provides an optimal description of TFBS statistics within statistical noise. The PIM generalizes previous approaches to interdependent positions: it accounts for co-variation of two or more base pairs, and predicts secondary motifs, while outperforming multiple-motif models consisting of mixtures of PWMs. -
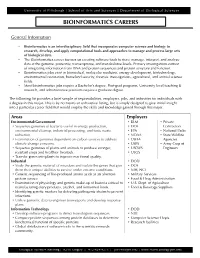
Bioinformatics Careers
University of Pittsburgh | School of Arts and Sciences | Department of Biological Sciences BIOINFORMATICS CAREERS General Information • Bioinformatics is an interdisciplinary field that incorporates computer science and biology to research, develop, and apply computational tools and approaches to manage and process large sets of biological data. • The Bioinformatics career focuses on creating software tools to store, manage, interpret, and analyze data at the genome, proteome, transcriptome, and metabalome levels. Primary investigations consist of integrating information from DNA and protein sequences and protein structure and function. • Bioinformatics jobs exist in biomedical, molecular medicine, energy development, biotechnology, environmental restoration, homeland security, forensic investigations, agricultural, and animal science fields. • Most bioinformatics jobs require a Bachelor’s degree. Post-grad programs, University level teaching & research, and administrative positions require a graduate degree. The following list provides a brief sample of responsibilities, employers, jobs, and industries for individuals with a degree in this major. This is by no means an exhaustive listing, but is simply designed to give initial insight into a particular career field that would employ the skills and knowledge gained through this major. Areas Employers Environmental/Government • BLM • Private • Sequence genomes of bacteria useful in energy production, • DOE Contractors environmental cleanup, industrial processing, and toxic waste • EPA • National Parks reduction. • NOAA • State Wildlife • Examination of genomes dependent on carbon sources to address • USDA Agencies climate change concerns. • USFS • Army Corp of • Sequence genomes of plants and animals to produce stronger, • USFWS Engineers resistant crops and healthier livestock. • USGS • Transfer genes into plants to improve nutritional quality. Industrial • DOD • Study the genetic material of microbes and isolate the genes that give • DOE them their unique abilities to survive under extreme conditions. -

Problems for Structure Learning: Aggregation and Computational Complexity
(in press). In S. Das, D. Caragea, W. H. Hsu, & S. M. Welch (Eds.), Computational methodologies in gene regulatory networks. Hershey, PA: IGI Global Publishing. Problems for Structure Learning: Aggregation and Computational Complexity Frank Wimberly Carnegie Mellon University (retired), USA David Danks Carnegie Mellon University and Institute for Human & Machine Cognition, USA Clark Glymour Carnegie Mellon University and Institute for Human & Machine Cognition, USA Tianjiao Chu University of Pittsburgh, USA ABSTRACT Machine learning methods to find graphical models of genetic regulatory networks from cDNA microarray data have become increasingly popular in recent years. We provide three reasons to question the reliability of such methods: (1) a major theoretical challenge to any method using conditional independence relations; (2) a simulation study using realistic data that confirms the importance of the theoretical challenge; and (3) an analysis of the computational complexity of algorithms that avoid this theoretical challenge. We have no proof that one cannot possibly learn the structure of a genetic regulatory network from microarray data alone, nor do we think that such a proof is likely. However, the combination of (i) fundamental challenges from theory, (ii) practical evidence that those challenges arise in realistic data, and (iii) the difficulty of avoiding those challenges leads us to conclude that it is unlikely that current microarray technology will ever be successfully applied to this structure learning problem. INTRODUCTION An important goal of cell biology is to understand the network of dependencies through which genes in a tissue type regulate the synthesis and concentrations of protein species. A mediating step in such synthesis is the production of messenger RNA (mRNA). -

A New Sequence Logo Plot to Highlight Enrichment and Depletion
bioRxiv preprint doi: https://doi.org/10.1101/226597; this version posted November 29, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. A new sequence logo plot to highlight enrichment and depletion. Kushal K. Dey 1, Dongyue Xie 1, Matthew Stephens 1, 2 1 Department of Statistics, University of Chicago 2 Department of Human Genetics, University of Chicago * Corresponding Email : [email protected] Abstract Background : Sequence logo plots have become a standard graphical tool for visualizing sequence motifs in DNA, RNA or protein sequences. However standard logo plots primarily highlight enrichment of symbols, and may fail to highlight interesting depletions. Current alternatives that try to highlight depletion often produce visually cluttered logos. Results : We introduce a new sequence logo plot, the EDLogo plot, that highlights both enrichment and depletion, while minimizing visual clutter. We provide an easy-to-use and highly customizable R package Logolas to produce a range of logo plots, including EDLogo plots. This software also allows elements in the logo plot to be strings of characters, rather than a single character, extending the range of applications beyond the usual DNA, RNA or protein sequences. We illustrate our methods and software on applications to transcription factor binding site motifs, protein sequence alignments and cancer mutation signature profiles. Conclusion : Our new EDLogo plots, and flexible software implementation, can help data analysts visualize both enrichment and depletion of characters (DNA sequence bases, amino acids, etc) across a wide range of applications.