Preferential Ribosome Loading on the Stress-Upregulated Mrna Pool Shapes the Selective Translation Under Stress Conditions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Polysome-Profiling in Small Tissue Samples
bioRxiv preprint doi: https://doi.org/10.1101/104596; this version posted February 1, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Polysome-profiling in small tissue samples Shuo Liang1,#, Hermano Bellato2,#, Julie Lorent1,#, Fernanda Lupinacci2, Vincent Van Hoef1, Laia Masvidal1,*, Glaucia Hajj2,* and Ola Larsson1,* 1. Department of Oncology-Pathology, Karolinska Institutet, Stockholm, 171 77, Sweden 2. International Research Center, A.C. Camargo Cancer Center, São Paulo, Brazil # Equally contributing authors * Correspondence: Laia Masvidal ([email protected]), Glaucia Hajj ([email protected]) and Ola Larsson ([email protected]) ABSTRACT Polysome-profiling is commonly used to study genome wide patterns of translational efficiency, i.e. the translatome. The standard approach for collecting efficiently translated polysome-associated RNA results in laborious extraction of RNA from a large volume spread across multiple fractions. This property makes polysome-profiling inconvenient for larger experimental designs or samples with low RNA amounts such as primary cells or frozen tissues. To address this we optimized a non-linear sucrose gradient which reproducibly enriches for mRNAs associated with >3 ribosomes in only one or two fractions, thereby reducing sample handling 5-10 fold. The technique can be applied to cells and frozen tissue samples from biobanks, and generates RNA with a quality reflecting the starting material. When coupled with smart-seq2, a single-cell RNA sequencing technique, translatomes from small tissue samples can be obtained. Translatomes acquired using optimized non-linear gradients are very similar to those obtained when applying linear gradients. -

Using Ribosome Profiling to Quantify Differences in Protein Expression
bioRxiv preprint doi: https://doi.org/10.1101/501478; this version posted December 19, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 Using ribosome profiling to quantify differences in protein expression: 2 a case study in Saccharomyces cerevisiae oxidative stress conditions 3 4 5 William R. Blevins1,#, Teresa Tavella1,#, Simone G. Moro1, Bernat Blasco-Moreno2, 6 Adrià Closa-Mosquera2, Juana Díez2, Lucas B. Carey2, M. Mar Albà1,2,3,* 7 8 1Evolutionary Genomics Groups, Research Programme on Biomedical Informatics (GRIB), Hospital del 9 Mar Research Institute (IMIM), Barcelona, Spain 10 2Health and Experimental Sciences Department, Universitat Pompeu Fabra(UPF), Barcelona, Spain 11 3Catalan Institution for Research and Advanced Studies (ICREA), Barcelona, Spain. 12 #Shared first co-authorship 13 *To whom correspondence should be addressed. 14 Running title: differential gene translation 15 Keywords: differential gene expression, ribosome profiling, RNA-Seq, translation, oxidative stress 1 bioRxiv preprint doi: https://doi.org/10.1101/501478; this version posted December 19, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 16 Abstract 17 18 Cells respond to changes in the environment by modifying the concentration of specific 19 proteins. Paradoxically, the cellular response is usually examined by measuring variations 20 in transcript abundance by high throughput RNA sequencing (RNA-Seq), instead of 21 directly measuring protein concentrations. -

Super-Resolution Ribosome Profiling Reveals Unannotated Translation Events in Arabidopsis
Super-resolution ribosome profiling reveals unannotated translation events in Arabidopsis Polly Yingshan Hsua, Lorenzo Calviellob,c, Hsin-Yen Larry Wud,1, Fay-Wei Lia,e,f,1, Carl J. Rothfelse,f, Uwe Ohlerb,c, and Philip N. Benfeya,g,2 aDepartment of Biology, Duke University, Durham, NC 27708; bBerlin Institute for Medical Systems Biology, Max Delbrück Center for Molecular Medicine, 13125 Berlin, Germany; cDepartment of Biology, Humboldt Universität zu Berlin, 10099 Berlin, Germany; dBioinformatics Research Center and Department of Statistics, North Carolina State University, Raleigh, NC 27695; eUniversity Herbarium, University of California, Berkeley, CA 94720; fDepartment of Integrative Biology, University of California, Berkeley, CA 94720; and gHoward Hughes Medical Institute, Duke University, Durham, NC 27708 Contributed by Philip N. Benfey, September 13, 2016 (sent for review June 30, 2016; reviewed by Pam J. Green and Albrecht G. von Arnim) Deep sequencing of ribosome footprints (ribosome profiling) maps and contaminants. Several metrics associated with translation have and quantifies mRNA translation. Because ribosomes decode mRNA been exploited (11), for example, the following: (i)ribosomesre- every 3 nt, the periodic property of ribosome footprints could be lease after encountering a stop codon (9), (ii) local enrichment of used to identify novel translated ORFs. However, due to the limited footprints within the predicted ORF (4, 13), (iii) ribosome footprint resolution of existing methods, the 3-nt periodicity is observed length distribution (7), and (iv) 3-nt periodicity displayed by trans- mostly in a global analysis, but not in individual transcripts. Here, we lating ribosomes (2, 6, 10, 14, 15). Among these features, some work report a protocol applied to Arabidopsis that maps over 90% of the well in distinguishing groups of coding vs. -

Integrated Analyses of Translatome and Proteome Identify the Rules of Translation Selectivity in RPS14-Deficient Cells
ARTICLE Red Cell Biology and its Disorders Integrated analyses of translatome and Ferrata Storti Foundation proteome identify the rules of translation selectivity in RPS14-deficient cells Ismael Boussaid,1 Salomé Le Goff,1,2,* Célia Floquet,1,* Emilie-Fleur Gautier,1,3 Anna Raimbault,1 Pierre-Julien Viailly,4 Dina Al Dulaimi,1 Barbara Burroni,5 Isabelle Dusanter-Fourt,1 Isabelle Hatin,6 Patrick Mayeux,1,2,3,# Bertrand Cosson7,# and Michaela Fontenay1,2,3,4,8 Haematologica 2021 1 Université de Paris, Institut Cochin, CNRS UMR 8104, INSERM U1016, Paris; Volume 106(3):746-758 2Laboratoire d’Excellence du Globule Rouge GR-Ex, Université de Paris, Paris; 3Proteomic Platform 3P5, Université de Paris, Paris; 4Centre Henri-Becquerel, Institut de Recherche et d’Innovation Biomedicale de Haute Normandie, INSERM U1245, Rouen; 5Assistance Publique-Hôpitaux de Paris, Centre-Université de Paris - Cochin, Service de Pathologie, Paris; 6Institute for Integrative Biology of the Cell (I2BC), CEA, CNRS, Université de Paris-Sud, Université Paris-Saclay, Gif-sur-Yvette Cedex; 7Université de Paris, Epigenetics and Cell Fate, CNRS UMR 7216, Paris and 8Assistance Publique- Hôpitaux de Paris, Centre-Université de Paris - Hôpital Cochin, Service d’Hématologie Biologique, Paris, France *SLG and CF contributed equally to this work. #PM and BC contributed equally as co-senior authors. ABSTRACT n ribosomopathies, the Diamond-Blackfan anemia (DBA) or 5q- syn- drome, ribosomal protein (RP) genes are affected by mutation or deletion, Iresulting in bone marrow erythroid hypoplasia. Unbalanced production of ribosomal subunits leading to a limited ribosome cellular content regu- lates translation at the expense of the master erythroid transcription factor GATA1. -

Minireview: Novel Micropeptide Discovery by Proteomics and Deep Sequencing Methods
fgene-12-651485 May 6, 2021 Time: 11:28 # 1 MINI REVIEW published: 06 May 2021 doi: 10.3389/fgene.2021.651485 Minireview: Novel Micropeptide Discovery by Proteomics and Deep Sequencing Methods Ravi Tharakan1* and Akira Sawa2,3 1 National Institute on Aging, National Institutes of Health, Baltimore, MD, United States, 2 Departments of Psychiatry, Neuroscience, Biomedical Engineering, and Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore, MD, United States, 3 Department of Mental Health, Johns Hopkins Bloomberg School of Public Health, Baltimore, MD, United States A novel class of small proteins, called micropeptides, has recently been discovered in the genome. These proteins, which have been found to play important roles in many physiological and cellular systems, are shorter than 100 amino acids and were overlooked during previous genome annotations. Discovery and characterization of more micropeptides has been ongoing, often using -omics methods such as proteomics, RNA sequencing, and ribosome profiling. In this review, we survey the recent advances in the micropeptides field and describe the methodological and Edited by: conceptual challenges facing future micropeptide endeavors. Liangliang Sun, Keywords: micropeptides, miniproteins, proteogenomics, sORF, ribosome profiling, proteomics, genomics, RNA Michigan State University, sequencing United States Reviewed by: Yanbao Yu, INTRODUCTION J. Craig Venter Institute (Rockville), United States The sequencing and publication of complete genomic sequences of many organisms have aided Hongqiang Qin, the medical sciences greatly, allowing advances in both human genetics and the biology of human Dalian Institute of Chemical Physics, Chinese Academy of Sciences, China disease, as well as a greater understanding of the biology of human pathogens (Firth and Lipkin, 2013). -

Efficient Analysis of Mammalian Polysomes in Cells and Tissues
TOOLS AND RESOURCES Efficient analysis of mammalian polysomes in cells and tissues using Ribo Mega-SEC Harunori Yoshikawa1†, Mark Larance1,2†, Dylan J Harney2, Ramasubramanian Sundaramoorthy1, Tony Ly1,3, Tom Owen-Hughes1, Angus I Lamond1* 1Centre for Gene Regulation and Expression, School of Life Sciences, University of Dundee, Dundee, United Kingdom; 2Charles Perkins Centre, School of Life and Environmental Sciences, University of Sydney, Sydney, Australia; 3Wellcome Centre for Cell Biology, University of Edinburgh, Edinburgh, United Kingdom Abstract We describe Ribo Mega-SEC, a powerful approach for the separation and biochemical analysis of mammalian polysomes and ribosomal subunits using Size Exclusion Chromatography and uHPLC. Using extracts from either cells, or tissues, polysomes can be separated within 15 min from sample injection to fraction collection. Ribo Mega-SEC shows translating ribosomes exist predominantly in polysome complexes in human cell lines and mouse liver tissue. Changes in polysomes are easily quantified between treatments, such as the cellular response to amino acid starvation. Ribo Mega-SEC is shown to provide an efficient, convenient and highly reproducible method for studying functional translation complexes. We show that Ribo Mega-SEC is readily combined with high-throughput MS-based proteomics to characterize proteins associated with polysomes and ribosomal subunits. It also facilitates isolation of complexes for electron microscopy and structural studies. *For correspondence: DOI: https://doi.org/10.7554/eLife.36530.001 [email protected] †These authors contributed equally to this work Introduction Competing interests: The The ribosome is a large RNA-protein complex, comprising four ribosomal RNAs (rRNAs) and >80 authors declare that no ribosomal proteins (RPs), which coordinates mRNA-templated protein synthesis. -

Comparative Analysis Reveals Genomic Features of Stress
Comparative analysis reveals genomic features of PNAS PLUS stress-induced transcriptional readthrough Anna Vilborga,b,1, Niv Sabathc, Yuval Wieselc, Jenny Nathansa,b, Flonia Levy-Adamc, Therese A. Yarioa,b, Joan A. Steitza,b, and Reut Shalgic,1 aDepartment of Molecular Biophysics and Biochemistry, Boyer Center for Molecular Medicine, Yale University School of Medicine, New Haven, CT 06536; bHoward Hughes Medical Institute, Yale University School of Medicine, New Haven, CT 06536; and cDepartment of Biochemistry, Rappaport Faculty of Medicine, Technion–Israel Institute of Technology, Haifa 31096, Israel Edited by Jasper Rine, University of California, Berkeley, CA, and approved July 14, 2017 (received for review July 10, 2017) Transcription is a highly regulated process, and stress-induced degraded by exonucleases that access the unprotected 5′ end changes in gene transcription have been shown to play a major role generated by cleavage at the polyA site (12, 13). However, recent in stress responses and adaptation. Genome-wide studies reveal studies show that various stress and disease states, including prevalent transcription beyond known protein-coding gene loci, osmotic stress (10), HSV-1 infection (9), and renal carcinoma generating a variety of RNA classes, most of unknown function. (8), increase both the levels and length of transcripts mapping to One such class, termed downstream of gene-containing transcripts regions downstream of the cleavage and polyadenylation sites. (DoGs), was reported to result from transcriptional readthrough Ourpreviousstudy(10)showedthat these transcripts are contin- upon osmotic stress in human cells. However, how widespread the uous with the RNAs generated from the upstream protein-coding readthrough phenomenon is, and what its causes and consequences gene, suggesting that they result from alterations in cleavage and are, remain elusive. -

Riboviz 2: a Flexible and Robust Ribosome Profiling Data Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2021.05.14.443910; this version posted May 17, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY 4.0 International license. i “RiboViz2_paper” — 2021/5/14 — 15:13 — page 1 — #1 i i i Application Note riboviz 2: A flexible and robust ribosome profiling data analysis and visualization workflow Alexander L. Cope 1, Felicity Anderson 2, John Favate 1, Michael Jackson 3, Amanda Mok 4, Anna Kurowska 2, Emma MacKenzie 2, Vikram Shivakumar 5, Peter Tilton 1, Sophie M. Winterbourne 2, Siyin Xue 2, Kostas Kavoussanakis 3, Liana F. Lareau 4,5, Premal Shah 1, Edward W.J. Wallace 2, 1Rutgers University, Department of Genetics, Piscataway, NJ, USA 2Institute for Cell Biology and SynthSys, School of Biological Sciences, The University of Edinburgh, Edinburgh, UK 3EPCC, The University of Edinburgh, Edinburgh, UK 4Center for Computational Biology, University of California, Berkeley, CA, USA 5Department of Bioengineering, University of California, Berkeley, CA, USA To whom correspondence should be addressed. E-mail: [email protected]; [email protected]; [email protected] Abstract Motivation: Ribosome profiling, or Ribo-seq, is the state of the art method for quantifying protein synthesis in living cells. Computational analysis of Ribo-seq data remains challenging due to the complexity of the procedure, as well as variations introduced for specific organisms or specialized analyses. Many bioinformatic pipelines have been developed, but these pipelines have key limitations in terms of functionality or usability. -

Improving Bacterial Ribosome Profiling Data Quality
bioRxiv preprint doi: https://doi.org/10.1101/863266; this version posted December 3, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Improving Bacterial Ribosome Profiling Data Quality Alina Glaub1, Christopher Huptas1, Klaus Neuhaus1,2, Zachary Ardern1* 1Chair for Microbial Ecology, Technical University of Munich, Freising, Germany, 2Core Facility Microbiome/NGS, Institute for Food & Health, Technical University of Munich, Freising, Germany *corresponding author Abstract Ribosome profiling (RIBO-seq) in prokaryotes has the potential to facilitate accurate detection of translation initiation sites, to increase understanding of translational dynamics, and has already allowed detection of many unannotated genes. However, protocols for ribosome profiling and corresponding data analysis are not yet standardized. To better understand the influencing factors, we analysed 48 ribosome profiling samples from 9 studies on E. coli K12 grown in LB medium. We particularly investigated the size selection step in each experiment since the selection for ribosome-protected footprints (RPFs) has been performed at various read lengths. We suggest choosing a size range between 22-30 nucleotides in order to obtain protein-coding fragments. In order to use RIBO-seq data for improving gene annotation of weakly expressed genes, the total amount of reads mapping to protein-coding sequences and not rRNA or tRNA is important, but no consensus about the appropriate sequencing depth has been reached. Again, this causes significant variation between studies. Our analysis suggests that 20 million non rRNA/tRNA mapping reads are required for global detection of translated annotated genes. -

Computational Resources for Ribosome Profiling: from Database to Web Server and Software Hongwei Wang*, Yan Wang* and Zhi Xie
Briefings in Bioinformatics, 2017, 1–12 doi: 10.1093/bib/bbx093 Paper Computational resources for ribosome profiling: from database to Web server and software Hongwei Wang*, Yan Wang* and Zhi Xie Corresponding authors: Hongwei Wang, State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, Guangzhou, China. Tel.: þ86 20 87335131; E-mail: [email protected]; Zhi Xie, State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, Guangzhou, China. Tel.: þ86 20 87335131; E-mail: [email protected] *These authors contributed equally to the work as joint first authors. Abstract Ribosome profiling is emerging as a powerful technique that enables genome-wide investigation of in vivo translation at sub-codon resolution. The increasing application of ribosome profiling in recent years has achieved remarkable progress to- ward understanding the composition, regulation and mechanism of translation. This benefits from not only the awesome power of ribosome profiling but also an extensive range of computational resources available for ribosome profiling. At pre- sent, however, a comprehensive review on these resources is still lacking. Here, we survey the recent computational ad- vances guided by ribosome profiling, with a focus on databases, Web servers and software tools for storing, visualizing and analyzing ribosome profiling data. This review is intended to provide experimental and computational biologists with a ref- erence to make appropriate choices among existing resources for the question at hand. Key words: ribosome profiling; translational regulation; database; Web server; software Introduction sub-codon resolution [6, 7]. By precisely pinpointing ribosomes during translation, this technique has provided unprecedented Precise control of gene expression can be directly modulated insights into the complexity of translatome [8–12]. -
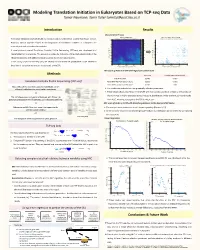
Modeling Translation Initiation in Eukaryotes Based on TCP-Seq Data Tamar Neumann, Tamir Tuller [email protected]
Modeling Translation Initiation in Eukaryotes Based on TCP-seq Data Tamar Neumann, Tamir Tuller [email protected] Introduction Results Discretization Process AUGsAUG's in in 5’UTR 5'UTR Signal Signal DiscretizationDiscretized Signal of AUG's- AUGs in 5'UTRin 5’UTR Signal Translation regulation and specifically its initiation step is fundamental to gene expression control. Highest 10% AUG Context Score Values Highest 10% AUG Context Score Values 100 3 85 However, various aspects related to the biophysics of translation initiation in eukaryotes are 2.8 85 2.6 75 currently not well understood or modeled. 75 2.4 65 A novel protocol named Translation Complex Profile Sequencing (TCP-seq) was developed and 65 2.2 55 55 2 implemented on S. cerevisiae. This protocol provides the footprints of the small subunit (SSU) of the 1.8 45 45 Footprint length [nt] length Footprint [nt] length Footprint Foot print length [nt] Foot print length [nt] 1.6 35 ribosome (possibly with additional factors) across the entire transcriptome. 35 1.4 25 25 In this study, based the TCP-seq data, we develop for the first-time quantitative model related to 1.2 15 1 15 the affect of transcript features on the dynamic of the SSU. -50 -25 AUG +25 +50 -50 -25 AUG +25 +50 Position relative to AUG Codon [nt] Position relative to AUG Codon [nt] MIC Score of AUGs in 5’UTR with High/Low Context Score Methods MIC Score P Value (1000 Permutations) AUG Start Codon 0.8795 <0.001 1 Translation Complex Profile Sequencing (TCP-seq) AUGs With High AUG Context Score 0.5230 <0.001 AUGs With Low AUG Context Score 0.1389 0.08 Yeast cells were crosslinked using formaldehyde, which Ø efficiently stabilizes the preinitiation complexes. -

Mrna-Seq and Ribosome Profiling Protocol Huili Guo, Bartel Lab, August 8, 2010
mRNA-Seq and Ribosome Profiling Protocol Huili Guo, Bartel lab, August 8, 2010 mRNA-Seq and Ribosome Profiling protocol Citation: Guo et al., Nature 466: 835‒840 (2010) This protocol describes the procedures used to perform mRNA-Seq and ribosome profiling on mammalian cells. In general, you would want to split each biological sample such that mRNA- Seq and ribosome profiling can be carried out in parallel for the same sample. If you are performing mRNA-Seq or ribosome profiling only for a given sample, skip to section 2 or 3, respectively. Structure In this protocol, some steps are specific to mRNA-Seq/ribosome profiling and indicated so. The library generation steps are largely common for both procedures. The last section includes steps for miscellaneous procedures used in the protocol. Section 1: Harvesting cells for parallel mRNA-Seq and ribosome profiling 1.1 Harvesting of cells/biological material The example below is written for cells grown in monolayer culture. (e.g. untransfected HeLa cells) Estimate of number of cells required: mRNA-Seq: Use enough cells to get ~15-20 μg total RNA Ribosome profiling: This varies depending on cell type/size, but as an example, I usually use ~10 million HeLa cells per sucrose gradient Day before: Split cells such that dish will be 70-80% confluent on day of harvesting. Day of harvesting: Arrest translation with cycloheximide (CHX) 1. Spike medium that cells are growing in with CHX (final 100 μg/ml, therefore NOTE the volume of media in the dish/container beforehand!) 2. Return cells to 37 °C for 10 min Harvesting cells 1.