AN ABSTRACT of the THESIS of NONG-KEUN SONG for the Degree
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Monocyclic Phenolic Acids; Hydroxy- and Polyhydroxybenzoic Acids: Occurrence and Recent Bioactivity Studies
Molecules 2010, 15, 7985-8005; doi:10.3390/molecules15117985 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Monocyclic Phenolic Acids; Hydroxy- and Polyhydroxybenzoic Acids: Occurrence and Recent Bioactivity Studies Shahriar Khadem * and Robin J. Marles Natural Health Products Directorate, Health Products and Food Branch, Health Canada, 2936 Baseline Road, Ottawa, Ontario K1A 0K9, Canada * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-613-954-7526; Fax: +1-613-954-1617. Received: 19 October 2010; in revised form: 3 November 2010 / Accepted: 4 November 2010 / Published: 8 November 2010 Abstract: Among the wide diversity of naturally occurring phenolic acids, at least 30 hydroxy- and polyhydroxybenzoic acids have been reported in the last 10 years to have biological activities. The chemical structures, natural occurrence throughout the plant, algal, bacterial, fungal and animal kingdoms, and recently described bioactivities of these phenolic and polyphenolic acids are reviewed to illustrate their wide distribution, biological and ecological importance, and potential as new leads for the development of pharmaceutical and agricultural products to improve human health and nutrition. Keywords: polyphenols; phenolic acids; hydroxybenzoic acids; natural occurrence; bioactivities 1. Introduction Phenolic compounds exist in most plant tissues as secondary metabolites, i.e. they are not essential for growth, development or reproduction but may play roles as antioxidants and in interactions between the plant and its biological environment. Phenolics are also important components of the human diet due to their potential antioxidant activity [1], their capacity to diminish oxidative stress- induced tissue damage resulted from chronic diseases [2], and their potentially important properties such as anticancer activities [3-5]. -

Influence of Whole-Wheat Consumption on Fecal Microbial Community
Influence of whole-wheat consumption on fecal microbial community structure of obese diabetic mice Jose F. Garcia-Mazcorro1,2, Ivan Ivanov3, David A. Mills4 and Giuliana Noratto5,# 1 Faculty of Veterinary Medicine, Universidad Auto´noma de Nuevo Leo´n, General Escobedo, Nuevo Leon, Mexico 2 Research Group Medical Eco-Biology, Universidad Auto´noma de Nuevo Leo´n, General Escobedo, Nuevo Leon, Mexico 3 Veterinary Physiology and Pharmacology, Texas A&M University, College Station, Texas, United States 4 Department of Food Science and Technology, University of California, Davis, Davis, California, United States 5 School of Food Science, Washington State University, Pullman, Washington, United States # Current Address: Nutrition and Food Science, Texas A&M University, College Station, Texas, United States ABSTRACT The digestive tract of mammals and other animals is colonized by trillions of metabolically-active microorganisms. Changes in the gut microbiota have been associated with obesity in both humans and laboratory animals. Dietary modifications can often modulate the obese gut microbial ecosystem towards a more healthy state. This phenomenon should preferably be studied using dietary ingredients that are relevant to human nutrition. This study was designed to evaluate the influence of whole-wheat, a food ingredient with several beneficial properties, on gut microorganisms of obese diabetic mice. Diabetic (db/db) mice were fed standard (obese-control) or whole-wheat isocaloric diets (WW group) for eight weeks; Submitted 28 October 2015 non-obese mice were used as control (lean-control). High-throughput sequencing Accepted 27 January 2016 using the MiSeq platform coupled with freely-available computational tools and Published 15 February 2016 quantitative real-time PCR were used to analyze fecal bacterial 16S rRNA gene Corresponding authors sequences. -

Methanogenesis Rates in Acetate and Nitrate Amended Anoxic Slurries
Methanogenesis rates in acetate and nitrate amended anoxic slurries BIOS 35502: Practicum in Environmental Field Biology Patrick Revord Advisor: William West 2011 Abstract With increasing urbanization and land use changes, pollution of lakes and wetland ecosystems is imminent. Any influx of nutrients, anthropogenic or natural, can have dramatic effects on lake gas production and flux. However, the net effect of simultaneous increase of both acetate and nitrate is unknown. Methane (CH4) production was measured in anoxic sediment and water slurries amended with ammonium nitrate (NH4NO3), which has been shown to inhibit methanogenesis, and sodium acetate (CH3COONa or NaOAc), which is known to increase methanogenesis. The addition of acetate significantly increased the methanogenesis rate, but the nitrate amendment had no significant effect. The simultaneous amendment of both acetate and nitrate showed no significant increase in CH4 compared to the control, indicating that the presence of nitrate may have reduced the effect of acetate amendment. Introduction Methane, a greenhouse gas associated with global warming, continues to increase in concentration in our atmosphere. Global yearly flux of methane into the atmosphere is 566 teragrams of CH4 per year, which is more than double pre-industrial yearly flux (Solomon et al. 2007). Increasing urbanization and land-use changes contribute significantly to increased gas levels (Anderson et al. 2010, Vitousek 1994). Nutrients travel from anthropogenic sources such as wastewater treatment facilities, landfills, and agricultural plots into nearby lakes, rivers, and wetlands, causing increased primary productivity in a process known as eutrophication (Vitousek et al. 1997). The increased nutrients and productivity lead to toxic algal blooms that create products such as acetate, H2, and CO2; a nutrient-rich anoxic environment suitable for anaerobic bacteria to produce unnaturally high levels of methane and other greenhouse gases (Davis and Koop 2006, West unpublished data). -

Hydroxybenzoic Acid Isomers and the Cardiovascular System Bernhard HJ Juurlink1,2, Haya J Azouz1, Alaa MZ Aldalati1, Basmah MH Altinawi1 and Paul Ganguly1,3*
Juurlink et al. Nutrition Journal 2014, 13:63 http://www.nutritionj.com/content/13/1/63 REVIEW Open Access Hydroxybenzoic acid isomers and the cardiovascular system Bernhard HJ Juurlink1,2, Haya J Azouz1, Alaa MZ Aldalati1, Basmah MH AlTinawi1 and Paul Ganguly1,3* Abstract Today we are beginning to understand how phytochemicals can influence metabolism, cellular signaling and gene expression. The hydroxybenzoic acids are related to salicylic acid and salicin, the first compounds isolated that have a pharmacological activity. In this review we examine how a number of hydroxyphenolics have the potential to ameliorate cardiovascular problems related to aging such as hypertension, atherosclerosis and dyslipidemia. The compounds focused upon include 2,3-dihydroxybenzoic acid (Pyrocatechuic acid), 2,5-dihydroxybenzoic acid (Gentisic acid), 3,4-dihydroxybenzoic acid (Protocatechuic acid), 3,5-dihydroxybenzoic acid (α-Resorcylic acid) and 3-monohydroxybenzoic acid. The latter two compounds activate the hydroxycarboxylic acid receptors with a consequence there is a reduction in adipocyte lipolysis with potential improvements of blood lipid profiles. Several of the other compounds can activate the Nrf2 signaling pathway that increases the expression of antioxidant enzymes, thereby decreasing oxidative stress and associated problems such as endothelial dysfunction that leads to hypertension as well as decreasing generalized inflammation that can lead to problems such as atherosclerosis. It has been known for many years that increased consumption of fruits and vegetables promotes health. We are beginning to understand how specific phytochemicals are responsible for such therapeutic effects. Hippocrates’ dictum of ‘Let food be your medicine and medicine your food’ can now be experimentally tested and the results of such experiments will enhance the ability of nutritionists to devise specific health-promoting diets. -

NMR Chemical Shifts of Common Laboratory Solvents As Trace Impurities
7512 J. Org. Chem. 1997, 62, 7512-7515 NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities Hugo E. Gottlieb,* Vadim Kotlyar, and Abraham Nudelman* Department of Chemistry, Bar-Ilan University, Ramat-Gan 52900, Israel Received June 27, 1997 In the course of the routine use of NMR as an aid for organic chemistry, a day-to-day problem is the identifica- tion of signals deriving from common contaminants (water, solvents, stabilizers, oils) in less-than-analyti- cally-pure samples. This data may be available in the literature, but the time involved in searching for it may be considerable. Another issue is the concentration dependence of chemical shifts (especially 1H); results obtained two or three decades ago usually refer to much Figure 1. Chemical shift of HDO as a function of tempera- more concentrated samples, and run at lower magnetic ture. fields, than today’s practice. 1 13 We therefore decided to collect H and C chemical dependent (vide infra). Also, any potential hydrogen- shifts of what are, in our experience, the most popular bond acceptor will tend to shift the water signal down- “extra peaks” in a variety of commonly used NMR field; this is particularly true for nonpolar solvents. In solvents, in the hope that this will be of assistance to contrast, in e.g. DMSO the water is already strongly the practicing chemist. hydrogen-bonded to the solvent, and solutes have only a negligible effect on its chemical shift. This is also true Experimental Section for D2O; the chemical shift of the residual HDO is very NMR spectra were taken in a Bruker DPX-300 instrument temperature-dependent (vide infra) but, maybe counter- (300.1 and 75.5 MHz for 1H and 13C, respectively). -
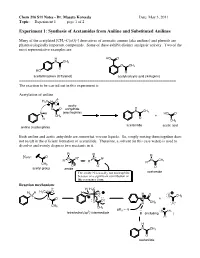
Experiment 1: Synthesis of Acetamides from Aniline and Substituted Anilines
Chem 216 S11 Notes - Dr. Masato Koreeda Date: May 3, 2011 Topic: __Experiment 1____ page 1 of 2. Experiment 1: Synthesis of Acetamides from Aniline and Substituted Anilines Many of the acetylated [CH3–C(=O)-] derivatives of aromatic amines (aka anilines) and phenols are pharmacologically important compounds. Some of these exhibit distinct analgesic activity. Two of the most representative examples are: H HO O N CH3 O CH3 O HO O acetaminophen (Tylenol) acetylsalicylic acid (Aspirin) ======================================================================= The reaction to be carried out in this experiment is: Acetylation of aniline δ- H3C δ+ O acetic O O anhydride H N CH3 N H (electrophile) HO O CH3 + H O CH3 acetanilide acetic acid aniline (nucleophile) Both aniline and acetic anhydride are somewhat viscous liquids. So, simply mixing them together does not result in the efficient formation of acetanilide. Therefore, a solvent (in this case water) is used to dissolve and evenly disperse two reactants in it. R R R Note: O + N R" N R" N CH R' R' R' 3 CH3 - O O O acetyl group amide The amide N is usually not nucleophilic acetamide because of a significant contribution of this resonance form. Reaction mechanism: δ- H H3C H H H3C δ+ O H N O H O N O CH3 O O N + O O H CH3 O CH3 CH3 pKa ~ -5 3 O CH3 tetrahedral (sp ) intermediate B (including ) O H N CH3 O acetanilide Chem 216 S11 Notes - Dr. Masato Koreeda Date: May 3, 2011 Topic: __Experiment 1____ page 2 of 2. Additional comments on the reaction mechanism: 1. -

The Genus Alpinia: a Review of Its Phytochemistry and Pharmacology
DOI: 10.15806/j.issn.2311-8571.2015.0026 World J Tradit Chin Med 2016; 2(1): 26–41 Modern Research on Chinese Materia Medica The Genus Alpinia: A Review of Its Phytochemistry and Pharmacology Wei-Jie Zhanga, Jian-Guang Luoa and Ling-Yi Kong* aState Key Laboratory of Natural Medicines, Department of Natural Medicinal Chemistry, China Pharmaceutical University, 24 Tong Jia Xiang, Nanjing 210009, China *Correspondence: Prof. Ling-Yi Kong, Department of Natural Medicinal Chemistry,China Pharmaceutical University,24 Tong Jia Xiang, Nanjing 210009, China, E-mail: [email protected] ABSTRACT Genus Alpinia consists of over 250 species, which are widely distributed in south and southeast Asia. Many plants of genus Alpinia have been used for thousands of years to treat digestive system diseases and as anti-inflammatory drugs. Phytochemical research on this genus has led to the isolation of different kinds of diarylheptanoids, terpenes triterpenoids, phenylbutanoids, lignans, and flavonoids. Experimental evidences revealed that both the crude extracts and pure constituents isolated from the genus Alpinia exhibit a wide range of bioactivities such as anti- cancer, anti-oxidant, anti-bacterial, anti-viral, cardiovascular, and digestive system protective effects. Here, we summarize the phytochemistry and pharmacology investigation of the genus Alpinia, which can provide reference for further research and drug development. Key words: Genus Alipinia, phytochemistry, pharmacology, a review Received 3 August 2015; Accept 2 March 2016 INTRODUCTION review, the conclusion can be drawn that, diarylheptanoids, terpenes and flavonoids are abundant in this genus. Genus Alpinia is a large genus of the Zingiberaceae family, which is widely distributed in many tropical regions of Asia, including China, India and Indonesia. -

Molecular Docking Study on Several Benzoic Acid Derivatives Against SARS-Cov-2
molecules Article Molecular Docking Study on Several Benzoic Acid Derivatives against SARS-CoV-2 Amalia Stefaniu *, Lucia Pirvu * , Bujor Albu and Lucia Pintilie National Institute for Chemical-Pharmaceutical Research and Development, 112 Vitan Av., 031299 Bucharest, Romania; [email protected] (B.A.); [email protected] (L.P.) * Correspondence: [email protected] (A.S.); [email protected] (L.P.) Academic Editors: Giovanni Ribaudo and Laura Orian Received: 15 November 2020; Accepted: 1 December 2020; Published: 10 December 2020 Abstract: Several derivatives of benzoic acid and semisynthetic alkyl gallates were investigated by an in silico approach to evaluate their potential antiviral activity against SARS-CoV-2 main protease. Molecular docking studies were used to predict their binding affinity and interactions with amino acids residues from the active binding site of SARS-CoV-2 main protease, compared to boceprevir. Deep structural insights and quantum chemical reactivity analysis according to Koopmans’ theorem, as a result of density functional theory (DFT) computations, are reported. Additionally, drug-likeness assessment in terms of Lipinski’s and Weber’s rules for pharmaceutical candidates, is provided. The outcomes of docking and key molecular descriptors and properties were forward analyzed by the statistical approach of principal component analysis (PCA) to identify the degree of their correlation. The obtained results suggest two promising candidates for future drug development to fight against the coronavirus infection. Keywords: SARS-CoV-2; benzoic acid derivatives; gallic acid; molecular docking; reactivity parameters 1. Introduction Severe acute respiratory syndrome coronavirus 2 is an international health matter. Previously unheard research efforts to discover specific treatments are in progress worldwide. -

Sodium Acetate
SODIUM ACETATE Prepared at the 18th JECFA (1974), published in NMRS 54B (1975) and in FNP 52 (1992). Metals and arsenic specifications revised at the 59th JECFA (2002). An ADI not limited' was established at the 17th JECFA (1973) SYNONYMS INS No. 262(i) DEFINITION Chemical names Sodium acetate C.A.S. number 127-09-3 Chemical formula C2H3NaO2 · nH2O (n = 0 or 3) Structural formula CH3COONa · nH2O (n = 0 or 3) Formula weight Anhydrous: 82.03 Trihydrate: 136.08 Assay Not less than 98.5% after drying DESCRIPTION Anhydrous: White, odourless, granular, hygroscopic powder Trihydrate: Colourless, transparent crystals or a granular crystalline powder, odourless or with a faint, acetic odour. Effloresces in warm, dry air. FUNCTIONAL USES Buffer CHARACTERISTICS IDENTIFICATION Solubility (Vol. 4) Very soluble in water; soluble in ethanol pH (Vol. 4) 8.0 - 9.5 (1 in 100 soln) Test for sodium (Vol. 4) Passes test Test for acetate (Vol. 4) Passes test Heat test Anhydrous: When heating the sample slowly, it first fuses gradually and boils, and later decomposes evolving an unpleasant odour of acetone. A solution of the residue gives alkaline reaction with litmus paper. Trihydrate: When heating the sample slowly, it liquefies. Then water evaporates, and a powder forms. By heating more strongly, the powder fuses, and becomes lumpy and later decomposes evolving an odour of acetone. A solution of the residue gives alkaline reaction with litmus paper. PURITY Loss on drying (Vol. 4) Anhydrous: Not more than 2.0% (120o, 4 h) Trihydrate: Between 36 and 42% (120o, 4 h) Test for potassium Negative test (Vol. -

Fitoterapia 145 (2020) 104610
Fitoterapia 145 (2020) 104610 Contents lists available at ScienceDirect Fitoterapia journal homepage: www.elsevier.com/locate/fitote Flavonoid, stilbene and diarylheptanoid constituents of Persicaria maculosa T Gray and cytotoxic activity of the isolated compounds Andrea Vasasa, Ildikó Lajtera, Norbert Kúsza, Péter Forgóa, Gusztáv Jakabb, Csilla Fazakasc, ⁎ Imola Wilhelmc,d, István A. Krizbaic,d, Judit Hohmanna,e, a Department of Pharmacognosy, University of Szeged, H-6720 Szeged, Hungary b Institute of Environmental Sciences, Faculty of Agricultural Water and Environmental Management, Tessedik Samuel College, H-5540 Szarvas, Hungary c Institute of Biophysics, Biological Research Centre, H-6726 Szeged, Hungary d Institute of Life Sciences, Vasile Goldiş Western University of Arad, RO-310414 Arad, Romania e Interdisciplinary Centre of Natural Products, University of Szeged, H-6720 Szeged, Hungary ARTICLE INFO ABSTRACT Keywords: Persicaria maculosa (Polygonaceae) has been used as edible and as medicinal plant since ancient times. As a result Persicaria maculosa of multistep chromatographic purifications, chalcones [2′-hydroxy-3′,4′,6′-trimethoxychalcone (1), pashanone Flavanones (2), pinostrobin chalcone (3)], flavanones [6-hydroxy-5,7-dimethoxyflavanone (4), pinostrobin (5), onysilin (6), Chalcones 5-hydroxy-7,8-dimethoxyflavanone (7)], flavonol [3-O-methylgalangin (8)], stilbene [persilben (9)], dia- Stilbene rylheptanoids [1,7-diphenylhept-4-en-3-one (10), dihydroyashabushiketol (12), yashabushidiol B (13)] and 3- Diarylheptanoids oxo-α-ionol-glucoside (11) were isolated from P. maculosa. The present paper reports for the first time the Cytotoxicity occurrence of diarylheptanoid-type constituents in the family Polygonaceae. Cytotoxicity of 1–5, 7 and 9–11 on 4 T1 mouse triple negative breast cancer cells was assayed by MTT test. -

Synthesis of Natural and Non-Natural Diarylheptanoids and Evaluation of Their Neuroprotective Activity
Synthesis of natural and non-natural diarylheptanoids and evaluation of their neuroprotective activity Dissertation zur Erlangung des Doktorgrades der Naturwissenschaften (Dr. rer. nat.) der Fakultät für Chemie und Pharmazie der Universität Regensburg vorgelegt von Petr Jirásek aus Prag 2014 Gedruckt mit Unterstützung des Deutschen Akademischen Austauschdienstes Die vorliegende Arbeit wurde im Zeitraum vom Januar 2011 bis September 2014 unter der Leitung von Prof. Dr. Jörg Heilmann und PD Dr. Sabine Amslinger am Lehrstuhl für Pharmazeutische Biologie und am Institut für Organische Chemie der Universität Regensburg angefertigt. Das Promotionsgesuch wurde eingereicht am: Tag der mündlichen Prüfung: 29.09.2014 Prüfungsausschuss: Prof. Dr. Jörg Heilmann (Erstgutachter) PD Dr. Sabine Amsliger (Zweitgutachter) Prof. Dr. Sigurd Elz (dritter Prüfer) Prof. Dr. Siavosh Mahboobi (Vorsitzender) Danksagung An dieser Stelle möchte ich mich bei allen Personen bedanken, die zum Gelingen dieser Arbeit beigetragen haben: bei Prof. Dr. Jörg Heilmann für die Vergabe dieses spannenden Themas, für viele wertvolle Anregungen, sein Vertrauen und für die schöne Jahre in seiner Arbeitsgruppe; bei PD Dr. Sabine Amslinger für die lehrreiche Zeit in Ihrem Umfeld, für all die Hilfe in Sachen organischer Chemie und für die herzliche Aufnahme in Ihrem Arbeitskreis; mein besonderer Dank gilt dem Deutschen Akademischen Austauschdienst für die Finanzierung meiner Promotion; bei allen jetzigen und ehemaligen Kolleginen und Kollegen am Lehrstuhl für Pharmazeutische Biologie möchte ich mich für das besonders freundliche Arbeitsklima und ihre Hilfsbereitschaft ganz herzlich bedanken; mein Dank gilt natürlich auch meinen Laborkolleginen und Laborkollegen von AK Amslinger für die angenehme Laborzeit und viele anregende Disskusionen; bei Frau Gabriele Brunner für ihre Geduld und Hilfe bei allen praktischen Aspekten der alltägigen Laborarbeit, genauso wie bei Frau Anne Grashuber für ihre unabdingbare und freundliche Unterstützung bei der Betreung der Praktika; weiterhin möchte ich mich bei Dr. -

3,4-DIHYDROXYBENZOIC ACID and 3,4-DIHYDROXYBENZALDEHYDE from the FERN Trichomanes Chinense L.; ISOLATION, ANTIMICROBIAL and ANTIOXIDANT PROPERTIES
Indo. J. Chem., 2012, 12 (3), 273 - 278 273 3,4-DIHYDROXYBENZOIC ACID AND 3,4-DIHYDROXYBENZALDEHYDE FROM THE FERN Trichomanes chinense L.; ISOLATION, ANTIMICROBIAL AND ANTIOXIDANT PROPERTIES Nova Syafni, Deddi Prima Putra, and Dayar Arbain* Faculty of Pharmacy/Sumatran Biota Laboratory, Andalas University, Kampus Limau Manis, Padang, 25163, West Sumatera, Indonesia Received May 1, 2012; Accepted September 5, 2012 ABSTRACT 3,4-dihydroxybenzoic acid (1) and 3,4-dihydroxybenzaldehyde (2) have been isolated from ethyl acetate fraction of methanolic fractions of leaves, stems and roots of the fern Trichomanes chinense L. (Hymenophyllaceae). These two compounds also showed significant antioxidant using DPPH and antimicrobial activities using the disc diffusion assay. Keywords: Trichomanes chinense L.; 3,4-dihydroxybenzoic acid; 3,4-dihydroxybenzaldehyde; antioxidant; antimicrobial ABSTRAK Telah diisolasi asam 3,4-dihidroksibenzoat (1) dan 3,4-dihidroksibenzaldehid (2) dari fraksi etil asetat ekstrak metanol daun, batang dan akar paku Trichomanes chinense L. (Hymenophyllaceae). Kedua senyawa ini memperlihatkan sifat antioksidan yang signifikan ketika diuji dengan metoda DPPH dan antimiokroba ketika diuji dengan metoda diffusi agar. Kata Kunci: Trichomanes chinense L.; asam 3,4-dihidroksibenzoat (1); 3,4-dihidroksibenzaldehid (2); antioksidan; antimikroba INTRODUCTION (Schizaeacee), Selaginella doederlinii Hieron, S. tamariscina (Bauv.) Spring, S. unsinata (Desv.) Spring. Based on a study on fossils particularly on that of (Sellaginellaceae) are also recorded to have medicinal Polypodiaceous family, it was considered that ferns have properties [7-8]. In continuation of our study on inhabited the earth as one of the pioneering plants since Sumatran ferns [6], chemical study, antimicrobial and the ancient time [1]. It was estimated that there were antioxidant properties of T.