Development of Specific Natural Bacterial Beta-Glucuronidase Inhibitors for Reducing Irinotecan-Associated Diarrhea
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Characterization of Prebiotics and Their Synergistic Activities with Lactobacillus Probiotics for Β-Glucuronidase Reduction
ESEARCH ARTICLE R ScienceAsia 45 (2019): 538–546 doi: 10.2306/scienceasia1513-1874.2019.45.538 Characterization of prebiotics and their synergistic activities with Lactobacillus probiotics for β-glucuronidase reduction a, a b a Achara Chaiongkarn ∗, Jirapa Dathong , Wipaporn Phatvej , Premsuda Saman , Chutima Kuanchaa, Lawan Chatanona, Somporn Moonmungmeea a Biodiversity Research Center, Thailand Institute of Scientific and Technological Research, Pathum Thani 12120 Thailand b Expert Center of Innovative Herbal Products, Thailand Institute of Scientific and Technological Research, Pathum Thani 12120 Thailand ∗Corresponding author, e-mail: [email protected] Received 1 Jun 2018 Accepted 28 Oct 2019 ABSTRACT: The role of synbiotics for enriching health and well-being in addition to suppressing disease is gaining interest. Synergistic activities of four candidate prebiotics as exopolysaccharides (EPSs) derived from Lactobacillus fer- mentum TISTR 2514 (EPS-TISTR 2514), Pediococcus acidilactici TISTR 2612 (EPS-TISTR 2612), manno-oligosaccharides and rice syrup-oligosaccharides were characterized and evaluated for decreasing the risk of colorectal cancer (CRC). Results revealed that one or more candidate prebiotics stimulated the growth of Lactobacillus casei DSM 20011, Lactobacillus plantarum DSM 2648, and Lactobacillus rhamnosus DSM 20021 by at least two orders of magnitude higher than positive control (using FOS as carbon source) within 24 h in vitro. Simulated gastrointestinal (pH 1) and α-amylase (pH 7) resistance were tested. Results showed more than 75% remaining after incubation at 37 °C after 6 h for all treatments except rice syrup. L. plantarum DSM 2648 + manno-oligosaccharides (Tr.1), L. plantarum DSM 2648 + EPS-TISTR 2612 (Tr.2), L. rhamnosus DSM 20021 + rice syrup-oligosaccharides (Tr.3), L. -

Isolated Co-Lipase Deficiency in Two Brothers
Gut: first published as 10.1136/gut.23.3.243 on 1 March 1982. Downloaded from Gut, 1982, 23, 243-246 Case reports Isolated co-lipase deficiency in two brothers H HILDEBRAND,* B BORGSTROM, A BEKASSY, C ERLANSON-ALBERTSSON, AND I HELIN From the Department ofPaediatrics and the Department ofPhysiological Chemistry, University ofLund, Sweden SUMMARY Two normally developed Assyrian brothers with isolated pancreatic co-lipase deficiency are described. They presented at the age of 5-6 years with loose stools. They had steatorrhoea, and analysis of exocrine pancreatic enzymes in the small intestine showed co-lipase deficiency, while amylase, chymotrypsin, trypsin, and lipase were normal. Intraduodenal infusion of purified co-lipase improved fat digestion measured by the triolein breath test. Their steatorrhoea diminished on treatment with enteric-coated pancreatic enzymes. The first indication for the existence of a co-factor for activities were assayed titrimetrically`5 using p-tosyl-l- pancreatic lipase was reported in 1963.1 In 1969 a arginine methyl ester (TAME) and N-acetyl-L-tyro- heat-stable co-factor was separated from lipase by gel- sine ethyl ester (ATEE), respectively, as substrates. filtration.2 Pure pancreatic lipase is inhibited by bile Lipase and co-lipase were measured titrimetrically salts in concentrations over their critical micellar con- using tributyrate as substrate.4 Total bile salt concen- centrations.3 The function of the co-factor, called co- trations and the ratio of glycine- to taurine-conjugated lipase, is to restore -

K113436 B. Purpose for Submi
510(k) SUBSTANTIAL EQUIVALENCE DETERMINATION DECISION SUMMARY ASSAY ONLY TEMPLATE A. 510(k) Number: k113436 B. Purpose for Submission: New device C. Measurand: Alkaline Phosphatase, Amylase, and Lactate Dehydrogenase D. Type of Test: Quantitative, enzymatic activity E. Applicant: Alfa Wassermann Diagnostic Technologies, LLC F. Proprietary and Established Names: ACE Alkaline Phosphatase Reagent Amylase Reagent ACE LDH-L Reagent G. Regulatory Information: Product Classification Regulation Section Panel Code CJE II 862.1050, Alkaline phosphatase 75-Chemistry or isoenzymes test system CIJ II 862.1070, Amylase test system 75-Chemistry CFJ II, exempt, meets 862.1440, Lactate 75-Chemistry limitations of dehydrogenase test system exemption. 21 CFR 862.9 (c) (4) and (9) H. Intended Use: 1. Intended use(s): See indications for use below. 2. Indication(s) for use: The ACE Alkaline Phosphatase Reagent is intended for the quantitative determination of alkaline phosphatase activity in serum using the ACE Axcel Clinical Chemistry System. Measurements of alkaline phosphatase are used in the diagnosis and treatment of liver, bone, parathyroid and intestinal diseases. This test is intended for use in clinical laboratories or physician office laboratories. For in vitro diagnostic use only. The ACE Amylase Reagent is intended for the quantitative determination α-amylase activity in serum using the ACE Axcel Clinical Chemistry System. Amylase measurements are used primarily for the diagnosis and treatment of pancreatitis (inflammation of the pancreas). This test is intended for use in clinical laboratories or physician office laboratories. For in vitro diagnostic use only. The ACE LDH-L Reagent is intended for the quantitative determination of lactate dehydrogenase activity in serum using the ACE Axcel Clinical Chemistry System. -

Extrapancreatic Sources of Lipase
Clinical Chemistry Byline David Parry, PhD, FCACB St. Boniface General Hospital James Dalton, PhD, FCACB Health Sciences Centre Extrapancreatic Sources of Lipase Reports now abound in the literature indicating that serum lipase is more specific and sensitive than amylase for detecting acute pancreatitis in patients presenting with acute abdominal pain (1-7). Amylase has been the mainstay biochemical test for the investigation of acute pancreatitis for many years, whereas lipase has not been widely used primarily because, until recently, reliable lipase assays were not suitable for rapid turnaround of results. Now, reliable and rapid lipase results are available at HSC, St. Boniface General Hospital, Seven Oaks General Hospital and Grace General Hospital. It is generally well appreciated that increases in serum amylase are seen not only in pancreatic disorders, but also in a number of non-pancreatic abdominal and non-abdominal diseases (1). With increased use of lipase, it has become increasingly recognized that lipase too can be elevated in clinical conditions that do not involve the pancreas. Even though the superiority of lipase over amylase for investigating acute pancreatitis has been shown repeatedly in recent literature (2-5), it is important to be aware that there are a number of clinical conditions that can give rise to elevated nonpancreatic lipase: 1) A recent study (2) has shown that some patients presenting with acute abdominal pain have elevations in serum lipase that are due to extrapancreatic disease processes. The sources of elevated lipase were attributed to a number of intra-abdominal anatomic locations other than the pancreas, including the biliary tract, esophagus, stomach, duodenum and small intestine. -

Diagnostic Value of Serum Enzymes-A Review on Laboratory Investigations
Review Article ISSN 2250-0480 VOL 5/ ISSUE 4/OCT 2015 DIAGNOSTIC VALUE OF SERUM ENZYMES-A REVIEW ON LABORATORY INVESTIGATIONS. 1VIDYA SAGAR, M.SC., 2DR. VANDANA BERRY, MD AND DR.ROHIT J. CHAUDHARY, MD 1Vice Principal, Institute of Allied Health Sciences, Christian Medical College, Ludhiana 2Professor & Ex-Head of Microbiology Christian Medical College, Ludhiana 3Assistant Professor Department of Biochemistry Christian Medical College, Ludhiana ABSTRACT Enzymes are produced intracellularly, and released into the plasma and body fluids, where their activities can be measured by their abilities to accelerate the particular chemical reactions they catalyze. But different serum enzymes are raised when different tissues are damaged. So serum enzyme determination can be used both to detect cellular damage and to suggest its location in situ. Some of the biochemical markers such as alanine aminotransferase, aspartate aminotransferase, alkaline phasphatase, gamma glutamyl transferase, nucleotidase, ceruloplasmin, alpha fetoprotein, amylase, lipase, creatine phosphokinase and lactate dehydrogenase are mentioned to evaluate diseases of liver, pancreas, skeletal muscle, bone, etc. Such enzyme test may assist the physician in diagnosis and treatment. KEYWORDS: Liver Function tests, Serum Amylase, Lipase, CPK and LDH. INTRODUCTION mitochondrial AST is seen in extensive tissue necrosis during myocardial infarction and also in chronic Liver diseases like liver tissue degeneration DIAGNOSTIC SERUM ENZYME and necrosis². But lesser amounts are found in Enzymes are very helpful in the diagnosis of brain, pancreas and lung. Although GPT is plentiful cardiac, hepatic, pancreatic, muscular, skeltal and in the liver and occurs only in the small amount in malignant disorders. Serum for all enzyme tests the other tissues. -

Pancreatic Amylase and Lipase Plasma Concentrations Are
Diabetes Care Volume 38, May 2015 e71 Pancreatic Amylase and Lipase Plasma David P. Sonne,1 Tina Vilsbøll,1 and Filip K. Knop1,2 Concentrations Are Unaffected by Increments in Endogenous GLP-1 Levels Following Liquid Meal Tests Diabetes Care 2015;38:e71–e72 | DOI: 10.2337/dc14-2751 Recent findings have suggested that amylase and lipase in plasma following patients with type 2 diabetes (P , incretin-based therapies promote pan- the ingestion of oral glucose and three 0.05), whereas lipase concentrations creatic inflammation and possibly cell isocaloric and isovolemic liquid mealsd were similar. Neither of the enzymes proliferation within the endocrine and all of which exerted normal endogenous increased following nutrient ingestion, exocrine pancreas (1). However, these GLP-1 secretion (4)din patients with suggesting that postprandial elevations studies have been met with substan- type 2 diabetes and matched control of endogenous GLP-1 (two- to three- tive criticism based on technical and subjects. fold) cannot trigger enzyme release methodical issues (2). Nevertheless, Detailed description of the experimen- from the human pancreas, at least not incretin-based therapies seem to result tal procedures and subjects was provided acutely. in small increases (within normal range) previously (4). In short, pancreas-specific These results suggest that the obser- in plasma concentrations of amylase amylase and lipase concentrations were vation of elevated plasma amylase and andlipaseinpatientsreceivingthese measured in plasma from 15 patients lipase -

Effect of Selenium Supplementation on Serum Amylase, Lactate Dehydrogenase and Alkaline Phosphatase Activities in Rats Exposed to Cadmium Or Lead
EFFECT OF SELENIUM SUPPLEMENTATION ON RATS EXPOSED TO CADMIUM OR LEAD Cercetări Agronomice în Moldova Vol. XLVII , No. 4 (160) / 2014 EFFECT OF SELENIUM SUPPLEMENTATION ON SERUM AMYLASE, LACTATE DEHYDROGENASE AND ALKALINE PHOSPHATASE ACTIVITIES IN RATS EXPOSED TO CADMIUM OR LEAD B.G. ŞLENCU1*, C. CIOBANU1, Carmen SOLCAN 2, Alina ANTON2, St. CIOBANU2, Gh. SOLCAN2, Rodica CUCIUREANU1 *E-mail: [email protected] Received June 13, 2014 ABSTRACT. The purpose of the study was Selenium, coadministered with cadmium, to assess the effect of selenium caused a marked increase in serum LDH supplementation on serum amylase, lactate activity, when compared to cadmium alone dehydrogenase (LDH) and alkaline or Control group while practically it had no phosphatase (ALP) activities in rats, during effect on lead induced changes in LDH subacute exposure to toxic doses of activity. Cadmium and lead induced cadmium or lead through the drinking disturbances in serum ALP activity were not water. The experimental groups (n=6) were: influenced by selenium supplementation. Control, Se (Se+4: 0,2 mg/l), Cd (Cd+2: 150 mg/l), Pb (Pb+2: 300 mg/l), Cd+Se (Cd+2: Key words: Rats; Lead; Cadmium; 150 mg/l; Se+4: 0,2 mg/l) and Pb+Se (Pb+2: Selenium; Blood serum enzyme. 300 mg/l; Se+4: 0,2 mg/l). The animals were sacrificed after 56 days. Amylase, LDH and REZUMAT. Efectul suplimentării cu ALP activities were determined from serum. seleniu asupra activităţii amilazei serice, Se and Pb treatments caused an increase in a lactat dehidrogenazei şi a fosfatazei amylase and LDH activities, when alcaline la sobolani, expuşi la cadmiu sau compared to Control group while Cd caused plumb. -

(PB) Is a Major Drug in the Treatment of Canine, Feline and Human Epilepsy and Can Significantly Reduce the Severity of Seizures
ACTA VET. BRNO 2002, 71: 309–312 Phenobarbital Effects on Brain and Liver Tissues Enzyme Activity in Balb/C Mice E. YAZAR 1*, O. DEMIR2, M. ELMAS1, A. L. BAS1, B. TRAS1 1Department of Pharmacology and Toxicology, Faculty of Veterinary Medicine, University of Selcuk, Konya, Turkey 2Department of Neurology, Faculty of Medicine, University of Selcuk, Konya, Turkey Received October 8, 2001 Accepted June 19, 2002 Abstract Yazar, E., O. Demir, M. Elmas, A. L. Bas, B. Tras: Phenobarbital Effects on Brain and Liver Tissues Enzyme Activity in Balb/C Mice. Acta Vet. Brno 71, 2002: 309-312. The purpose of the present study was to investigate the effect of phenobarbital on some brain and liver tissue enzyme activities in Balb/C mice. Forty male Balb/C mice were used. Ten mice served as a control group, and thirty mice were administered with phenobarbital (80 mg·kg-1 body mass, single oral dose). Brain and liver samples were taken at 6, 12 and 24 h after drug administration. Brain and liver tissues alkaline phosphatase, aspartate aminotransferase, gamma glutamyl transferase and amylase activities were measured by auto- analyzer. Phenobarbital did not affect gamma glutamyl transferase and amylase activities in the brain and liver, and alkaline phosphatase and aspartate aminotransferase activities in the liver. However, statistically significant (p < 0.05) increases of alkaline phosphatase and aspartate aminotransferase activities were observed in the brain. In general, after phenobarbital administration, serum alkaline phosphatase and aspartate aminotransferase activities increase and these increases are believed to be derived from the liver. These result suggest that brain may contribute to increased activities of these enzymes in serum in Balb/C mice. -

Beta-Amylase Action on High Molecular Weight Maltosaccharides Rudolph William Youngquist Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1962 Beta-amylase action on high molecular weight maltosaccharides Rudolph William Youngquist Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Biochemistry Commons Recommended Citation Youngquist, Rudolph William, "Beta-amylase action on high molecular weight maltosaccharides " (1962). Retrospective Theses and Dissertations. 2033. https://lib.dr.iastate.edu/rtd/2033 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. BETA-AMYLASE ACTION ON HIGH MOLECULAR WEIGHT MALTOSACCHARIDES by Rudolph William Youngqulst A Dissertation Submitted to the Graduate Faculty in Partial Fulfillment of The Requirements for the Degree of DOCTOR OF PHILOSOPHY Major Subject: Biochemistry Signature was redacted for privacy. In Charge of Major Work Signature was redacted for privacy. Head of Major Department Signature was redacted for privacy. Iowa State University Of Science and Technology Ames, Iowa 1962 11 TABLE OF CONTENTS Page INTRODUCTION . 1 LITERATURE REVIEW 3 Beta-amylase 3 Action pattern 4 Active site 7 Maltosaccharides 8 METHODS AND MATERIALS 10 Carbohydrates 10 Amylodextrin 10 Maltosaccharides 10 Amylase 10 Chromatographic Techniques 10 Analytical 11 Nelson' s test 12 Dinitrosalicylic acid test 12 Quantitative determinations 12 EXPERIMENTAL 14 Stability of Beta-amylase 14 Action Pattern of Beta-amylase on Maltosaccharides. 17 Enzyme activity 17 Digests. -

STUDY of STARCH DEBRANCHING ENZYMES in DEVELOPING WHEAT KERNELS a Thesis Submitted to the College of Graduate Studies and Resear
STUDY OF STARCH DEBRANCHING ENZYMES IN DEVELOPING WHEAT KERNELS A Thesis Submitted to the College of Graduate Studies and Research in Partial Fulfilment of the Requirements for the Degree of Doctor of Philosophy in the Department of Biochemistry University of Saskatchewan Saskatoon By Supatcharee Netrphan Spring 2002 © Copyright Supatcharee Netrphan, 2002. All rights reserved. PERMISSION TO USE In presenting this thesis in partial fulfilment of the requirements for a Postgraduate degree from the University of Saskatchewan, I agree that the Libraries of this University may make it freely available for inspection. I further agree that permission for copying of this thesis in any manner, in whole or in part, for scholarly purposes may be granted by the professor or professors who supervised my thesis work or, in their absence, by the Head of the Department or the Dean of the College in which my thesis work was done. It is understood that any copying or publication or use of this thesis or parts thereof for financial gain shall not be allowed without my written permission. It is also understood that due recognition shall be given to me and to the University of Saskatchewan in any scholarly use which may be made of any material in my thesis. Requests for permission to copy or to make other use of material in this thesis in whole or in part should be addressed to: Head of the Department of Biochemistry University of Saskatchewan 107 Wiggins Road Saskatoon, Saskatchewan S7N 5E5 i ABSTRACT Starch debranching enzymes, which specifically hydrolyse a-1,6 glucosidic bonds in glucans containing both a-1,4 and a-1,6 linkages, are classified into two types: isoamylase (EC. -

Beta-Amylase
www.megazyme.com BETA-AMYLASE ASSAY PROCEDURE (BETAMYL-3® METHOD) K-BETA3 12/19 (100/200 Assays per Kit) © Megazyme 2019 INTRODUCTION: β-Amylase plays a central role in the complete degradation of starch to metabolisable or fermentable sugars during the germination or malting of cereal grains. It also finds considerable application, together with starch debranching enzymes, in the production of high maltose syrups. β-Amylase is usually measured using non-specific reducing sugar assays with starch as substrate. In some methods, the α-amylase is first inactivated by treatment at low pH. A major advance in the assay of β-amylase was introduced by Mathewson and Seabourn1 who found that the Calbiochem Pantrak® serum α-amylase reagent could be used to measure β-amylase in the presence of cereal α-amylase. The reagent (Pantrak) consists of a mixture of p-nitrophenyl-α-D-maltopentaoside (PNPG5) and p-nitrophenyl-α-D-maltohexaoside (PNPG6). These substrates are rapidly hydrolysed by β-amylase, but are only slowly cleaved by cereal α-amylase, which requires a longer stretch of α-1,4-linked D-glucosyl residues to satisfy the substrate sub-site binding requirements. Subsequently, Megazyme offered a product, Betamyl® (β-Amylase Assay Reagent) that comprised just PNPG5 and α-glucosidase,2 which gave greater specificity. This reagent is now superceded by the Megazyme Betamyl-3®, β-Amylase assay reagent, which is more specific and considerably more stable than the Betamyl® reagent. The Megazyme Betamyl-3®, β-amylase test reagent employs high purity β-glucosidase and p-nitrophenyl-β-D-maltotrioside (PNPβ-G3). -
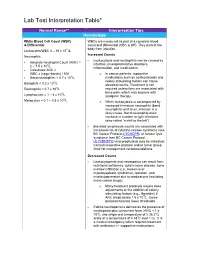
Lab Test Interpretation Table*
Lab Test Interpretation Table* Normal Range** Interpretation Tips Hematology White Blood Cell Count (WBC) WBCs are measured as part of a complete blood & Differential count and differential (CBC & diff). They protect the body from infection. Leukocytes/WBC 4 – 10 x 109 /L Increased Counts Neutrophils - Leukocytosis and neutrophilia can be caused by Absolute Neutrophil Count (ANC) = - infection, myeloproliferative disorders, 2 – 7.5 x 109/L inflammation, and medications. - Calculated ANC = WBC x (segs+bands) / 100 o In cancer patients, supportive - Band neutrophils: < 0.7 x 109/L medications such as corticosteroids and 9 colony stimulating factors can cause Basophils < 0.2 x 10 /L elevated counts. Treatment is not Eosinophils < 0.7 x 109/L required unless they are associated with 9 bone pain, which may improve with Lymphocytes = 1 – 4 x 10 /L analgesic therapy. 9 Monocytes = 0.1 – 0.8 x 10 /L o When leukocytosis is accompanied by increased immature neutrophils (band neutrophils) and fever, infection is a likely cause. Band neutrophils often increase in number to fight infections (also called “a shift to the left”). - Elevated lymphocyte counts are associated with increased risk of cytokine-release syndrome (see BC Cancer Protocol LYCHOPR) or tumour lysis syndrome (see BC Cancer Protocol ULYVENETO) and prophylaxis may be indicated. Consult respective protocol and/or tumor group chair for management recommendations. Decreased Counts - Leukocytopenia and neutropenia can result from nutritional deficiency, autoimmune disease, bone marrow infiltration (i.e., leukemia or myelodysplastic syndrome), radiation, and myelosuppression due to medications (including many cancer drugs). o Many treatment protocols require dose adjustments or the addition of colony stimulating factors (e.g., filgrastim) if ANC drops below 1.5 x 109/L.