Development and Characterization of Raffia Palm Fiber Reinforced Polymer Matrix Composites
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
UNITED STATES NATIONAL MUSEUM Bulletin 147
Q 11 U563 CRLSSI BULLETIN 147 MAP U. S. NATIONAL MUSEUM 's SMITHSONIAN INSTITUTION UNITED STATES NATIONAL MUSEUM Bulletin 147 ARCHEOLOGICAL AND HISTORICAL INVESTIGATIONS IN SAMANA DOMINICAN REPUBLIC BY HERBERT W. KRIEGER Curator of Ethnology, United States National Museum UNITED STATES GOVERNMENT PRINTING OFFICE WASHINGTON : 1929 For sale by ths Superintendent of Documents, Washington, D. C. Price 40 Cents ADVERTISEMENT The scientific publications of the National Museum include two series, known, respectively, as Proceedings and Bulletin. The Proceedings, begun in 1878, is intended primarily as a medium for the publication of original papers, based on the collections of the National Museum, that set forth newly acquired facts in biology, anthropology, and geology, with descriptions of new forms and revisions of limited groups. Copies of each paper, in pamphlet form, are distributed as published to libraries and scientific organ- izations and to specialists and others interested in the different subjects. The dates at which these separate papers are published are recorded in the table of contents of each of the volumes. The Bulletin, the first of which was issued in 1875, consist of a series of separate publications comprising monographs of large zoological groups and other general systematic treatises (occasion- ally in several volumes), faunal works, reports of expeditions, cata- logues of type-specimens, special collections, and other material of similar nature. The majority of the volumes are octavo in size, but a quarto size has been adopted in a few instances in which large plates were regarded as indispensable. In the Bulletin series appear volumes under the heading Contributions from the United States National Herharium, in octavo form, published by the National Museum since 1902, which contain papers relating to the botanical collections of the Museum. -

Evaluation of the Mechanical Properties on Sisal-Coir Hybrid Natural Fiber Composites
International Journal Of Engineering Research And Development e-ISSN: 2278-067X, p-ISSN: 2278-800X, www.ijerd.com Volume 13, Issue 9 (September 2017), PP.43-49 Evaluation of The Mechanical Properties on Sisal-Coir Hybrid Natural Fiber Composites * 1 2 3 Madhukiran.J , Dr.T.Venkateswara Rao ,Dr.S.Madhusudan , Dr. R.Umamaheswara Rao4 1Asst.Professor, Dept.of Mechanical Engineering, Sasi Institute of Technology and Engineering, Tadepalligudem,-534101, Andhra Pradesh, India. 2 Professor, Dept.of Mechanical Engineering, Bonam Venkata Chalamayya Engineering College, Odalarevu, Amalapuram, Andhra Pradesh-533210, India. 3Professor, Dept.of Mechanical Engineering, Usha Rama College of Engineering and Technology Telaprolu, Andhra Pradesh, India. 4Professor & Head, Dept.of Mechanical Engineering, Sasi Institute of Technology and Engineering, Tadepalligudem, Andhra Pradesh, India. Corresponding Author: *Madhukiran.J ABSTRACT: - The usage of natural fiber reinforced composites is growing at a faster rate and is actively being considered as an alternate material for synthetic fiber. In the present work an attempt has been made to fabricate the sisal-coir fiber randomly oriented hybrid composites and to evaluate the mechanical properties such as tensile strength and flexural strength. Three types (sisal-coir) of hybrid laminate and two pure laminate composites are fabricated using manual layup technique. Epoxy (Ly556 and HY951) resin is used as matrix material in the present work. The specimens are prepared according to ASTM standards and the experiments were conducted on an universal testing machine (UTM).From the experimental results, it has been observed that the sisal-coir fiber hybrid composites exhibited superior properties when compared to pure composites. Keywords:- Coir & Sisal fibers, Epoxy, Hybrid polymer matrix composite, Hand layup. -

Physico-Chemical and Nutritional Characteristics, and Antimicrobial Activity of Oil Palm Syrup, Raffia Palm Syrup and Honey
IOSR Journal of Pharmacy and Biological Sciences (IOSR-JPBS) e-ISSN: 2278-3008, p-ISSN:2319-7676. Volume 11, Issue 1 Ver. I (Jan. - Feb. 2016), PP 73-78 www.iosrjournals.org Physico-chemical and Nutritional Characteristics, and Antimicrobial Activity of Oil Palm Syrup, Raffia Palm Syrup and Honey *Fred Omon Oboh, Lorenta Iyare, Monday Idemudia and Stephen Enabulele. Department of Basic Sciences, Benson Idahosa University, P.M.B. 1100, Benin City, Nigeria. Correspondence e-mail: [email protected], [email protected] Abstract: The physico-chemical characteristics and antimicrobial activity of oil palm syrup, raffia palm syrup and honey were studied. The materials contained mainly carbohydrate (64.76-68.79%) and water (28.05- 31.50). They exhibited similar densities (1.23-1.26 g cm-3) and pH (3.51-4.18), and had low ash (0.30-0.50%), protein (0.24-1.04%) and lipid (2.20-3.62%) content. They had modest content of Fe (2.35-3.30 mg/100g), Ca (37.06-79.05 mg/100g), and phenolic compounds (125.93-185.44 mg GAE /100 g), and were rich in potassium (325.12-628.56 mg/100g). They contained non-enzymatic browning products (browning intensity was 0.71 for honey, 0.159 for raffia palm syrup and 0.175 for oil palm syrup). The materials exhibited antimicrobial activity against clinical strains of Pseudomonas aeruginosa, Bacillus cereus, Escherichia coli and Staphylococcus aureus. The antimicrobial activities of dilute solutions (0.1 ml, 0.5% aqueous solutions) of the honey (containing 360 µg dry matter) and syrups (raffia, 360 µg and oil palm, 340 µg dry matter) were similar to that of 10 µg of the antibiotic streptomycin. -

Identification of Callose Synthases in Stinging Nettle and Analysis Of
International Journal of Molecular Sciences Communication Identification of Callose Synthases in Stinging Nettle and Analysis of Their Expression in Different Tissues Gea Guerriero 1,* , Emilie Piasecki 1, Roberto Berni 2 , Xuan Xu 1, Sylvain Legay 1 and Jean-Francois Hausman 1 1 Environmental Research and Innovation (ERIN) Department, Luxembourg Institute of Science and Technology, 5, rue Bommel, Z.A.E. Robert Steichen, L-4940 Hautcharage, Luxembourg; [email protected] (E.P.); [email protected] (X.X.); [email protected] (S.L.); [email protected] (J.-F.H.) 2 Department of Life Sciences, University of Siena, via P.A. Mattioli 4, I-53100 Siena, Italy; [email protected] * Correspondence: [email protected]; Tel.: +352-275-888-5096; Fax: +352-275-8885 Received: 14 May 2020; Accepted: 27 May 2020; Published: 28 May 2020 Abstract: Callose is an important biopolymer of β-1,3-linked glucose units involved in different phases of plant development, reproduction and response to external stimuli. It is synthesized by glycosyltransferases (GTs) known as callose synthases (CalS) belonging to family 48 in the Carbohydrate-Active enZymes (CAZymes) database. These GTs are anchored to the plasma membrane via transmembrane domains. Several genes encoding CalS have been characterized in higher plants with 12 reported in the model organism Arabidopsis thaliana. Recently, the de novo transcriptome of a fibre-producing clone of stinging nettle (Urtica dioica L.) was published and here it is mined for CalS genes with the aim of identifying members differentially expressed in the core and cortical tissues of the stem. -

Cuba: Informe Nacional Sobre Los Rfaa
CUBA: INFORME NACIONAL SOBRE LOS RFAA La Habana, abril 2007 1 INDICE Página LISTA DE ACRÓNIMOS 4 SECCIÓN I: SUMARIO EJECUTIVO. 5 SECCIÓN II: INTRODUCCIÓN AL PAÍS Y AL SECTOR AGRÍCOLA. 9 II.1. Principales características de la naturaleza y la biodiversidad de Cuba. 9 II. 2. Características socio-económicas de Cuba. El Plan Turquino y su papel en la agricultura cubana. 10 II.3. Principales sistemas de producción, cultivos y productos animales, así como exportaciones. 11 SECCIÓN III. CUERPO PRINCIPAL DEL INFORME DE PAÍS. 15 Capítulo 1: Estado de la Diversidad. 15 1.1. El estado de la diversidad e importancia relativa para la seguridad alimentaria de granos básicos y cereales. 15 1.2. El estado de la diversidad e importancia relativa de raíces, tubérculos y rizomas, plátanos y bananos. 17 1.3. El estado de la diversidad e importancia relativa para la seguridad alimentaria de los frutales. 18 1.4. El estado de la diversidad e importancia relativa para la seguridad alimentaria en hortalizas y oleaginosas. 18 1.5. El estado de la diversidad en los cultivos industriales (caña de azúcar, café, cacao, tabaco y fibras). 20 1.6. El estado de la diversidad de los pastos y forrajes y plantas silvestres útiles. 21 1.7. El estado de la diversidad en los recursos genéticos forestales. 22 1.8. El estado de la diversidad en los cultivos subutilizados. 24 1.9. Consideraciones generales. 24 Capítulo 2: El Estado del Manejo In Situ de los RFAA. 25 2.1. Inventarios de la agrobiodiversidad in situ. 25 2.2. Apoyo para el ordenamiento en y mejoramiento en fincas de los 26 RFAA. -

Investigation of Mechanical Behaviour of Sisal Bamboo and Epoxy Reinforced Natural Composite
International Journal of Engineering Applied Sciences and Technology, 2021 Vol. 5, Issue 12, ISSN No. 2455-2143, Pages 307-310 Published Online April 2021 in IJEAST (http://www.ijeast.com) INVESTIGATION OF MECHANICAL BEHAVIOUR OF SISAL BAMBOO AND EPOXY REINFORCED NATURAL COMPOSITE Gobi K Kaleeswaran M Department of Mechanical Engineering Department of Mechanical Engineering P.A. College of Engineering and Technology, P.A. College of Engineering and Technology, Pollachi, Coimbatore, India. Pollachi, Coimbatore, India. Amaresh D Dhanush R Department of Mechanical Engineering Department of Mechanical Engineering P.A. College of Engineering and Technology, P.A. College of Engineering and Technology, Pollachi, Coimbatore, India. Pollachi, Coimbatore, India. Abstract - Now-a-days, The natural sisal/bamboos from serve the desirable properties. Further, though composite types renewable natural resources offer the potential to act as a are often distinguishable from one another, no clear reinforcing material for polymer composites alternative to determination can be really made. The demands on matrices are the use of glass, carbon and wood. Among various natural many. They may need to temperature variations, be conductors fibers, sisal or bamboo is most widely used natural fiber due or resistors of electricity, have moisture sensitivity etc. This to its advantages like easy availability, low density, low may offer weight advantages, ease of handling and other merits production cost and satisfactory mechanical properties. For which may also become applicable depending on the purpose a composite material, its mechanical behavior depends on for which matrices are chosen. Solids that accommodate stress many factors such as fiber content, orientation, types, to incorporate other constituents provide strong bonds for the length etc. -

Complete Sequence of Kenaf (Hibiscus Cannabinus)
www.nature.com/scientificreports OPEN Complete sequence of kenaf (Hibiscus cannabinus) mitochondrial genome and comparative analysis Received: 2 November 2017 Accepted: 27 July 2018 with the mitochondrial genomes of Published: xx xx xxxx other plants Xiaofang Liao1,2,3, Yanhong Zhao3, Xiangjun Kong2, Aziz Khan2, Bujin Zhou 2, Dongmei Liu4, Muhammad Haneef Kashif2, Peng Chen2, Hong Wang5 & Ruiyang Zhou2 Plant mitochondrial (mt) genomes are species specifc due to the vast of foreign DNA migration and frequent recombination of repeated sequences. Sequencing of the mt genome of kenaf (Hibiscus cannabinus) is essential for elucidating its evolutionary characteristics. In the present study, single- molecule real-time sequencing technology (SMRT) was used to sequence the complete mt genome of kenaf. Results showed that the complete kenaf mt genome was 569,915 bp long and consisted of 62 genes, including 36 protein-coding, 3 rRNA and 23 tRNA genes. Twenty-fve introns were found among nine of the 36 protein-coding genes, and fve introns were trans-spliced. A comparative analysis with other plant mt genomes showed that four syntenic gene clusters were conserved in all plant mtDNAs. Fifteen chloroplast-derived fragments were strongly associated with mt genes, including the intact sequences of the chloroplast genes psaA, ndhB and rps7. According to the plant mt genome evolution analysis, some ribosomal protein genes and succinate dehydrogenase genes were frequently lost during the evolution of angiosperms. Our data suggest that the kenaf mt genome retained evolutionarily conserved characteristics. Overall, the complete sequencing of the kenaf mt genome provides additional information and enhances our better understanding of mt genomic evolution across angiosperms. -

+Woven Furnishings Inspired Children's Rooms
connecticut cottages connecticut cottages & gardens april 2021 | COTTAGESGARDENS.COM APRIL 2021 cottagesgardens.com WOVEN FURNISHINGS FRESH TAKE INSPIRED CHILDREN’S ON DESIGN +ROOMS WHAT’S NEW Out of the Box WRAPPED AND WOVEN—ORGANIC FURNISHINGS IN WICKER, RAFFIA, RATTAN AND ROPE | PRODUCED BY MARY FITZGERALD HANGING AROUND Inside or out, the Marina hanging chair from Arhaus invites fun and relaxation. Constructed in a powder- coated aluminum, the frame is wrapped in an all-weather rope. $2,349, SoNo Collection, Norwalk, arhaus.com. SUNNY OUTLOOK The ornate flower motif of Made Good’s Waverly mirror is formed from woven natural rattan wicker. $1,350, available through Housewarmings, Greenwich, madegoods.com. SIDE BAR The Balabac rattan sideboard from British style brand Oka is fashioned from mindi and durian wood. Rattan-wrapped doors are accented with metal ring handles. $1,650, oka.com. april 2021 cottagesgardens.com ctc&g 27 WHAT’S NEW WHAT’S NEW ROARING ’20S This rattan single CRISSCROSS PATTERN drawer bedside table The macramé effect of the Bandelier hails from Soane’s accent chair from Longaberger is Templeton collection achieved with interlaced white cowhide and was inspired by leather. The Danish- a 1920s design. The inspired design is SCREEN TIME elegant curved sides of finely crafted with Made in France, pale oak are wrapped sungkai wood in a the Scale screen in rattan. $5,125, natural finish. $459, from Atelier Vime is soane.com. longaberger.com. woven in rattan with A NEW TWIST wood and leather. Finely twisted jute is stretched over the metal frame of The French word the Manhattan pendant light from Selamat. -

Outline of Angiosperm Phylogeny
Outline of angiosperm phylogeny: orders, families, and representative genera with emphasis on Oregon native plants Priscilla Spears December 2013 The following listing gives an introduction to the phylogenetic classification of the flowering plants that has emerged in recent decades, and which is based on nucleic acid sequences as well as morphological and developmental data. This listing emphasizes temperate families of the Northern Hemisphere and is meant as an overview with examples of Oregon native plants. It includes many exotic genera that are grown in Oregon as ornamentals plus other plants of interest worldwide. The genera that are Oregon natives are printed in a blue font. Genera that are exotics are shown in black, however genera in blue may also contain non-native species. Names separated by a slash are alternatives or else the nomenclature is in flux. When several genera have the same common name, the names are separated by commas. The order of the family names is from the linear listing of families in the APG III report. For further information, see the references on the last page. Basal Angiosperms (ANITA grade) Amborellales Amborellaceae, sole family, the earliest branch of flowering plants, a shrub native to New Caledonia – Amborella Nymphaeales Hydatellaceae – aquatics from Australasia, previously classified as a grass Cabombaceae (water shield – Brasenia, fanwort – Cabomba) Nymphaeaceae (water lilies – Nymphaea; pond lilies – Nuphar) Austrobaileyales Schisandraceae (wild sarsaparilla, star vine – Schisandra; Japanese -
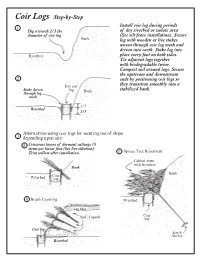
Coir Logs Step-By-Step Install Coir Log During Periods 1 Dig a Trench 2/3 the of Dry Riverbed Or Isolate Area Diameter of Coir Log (See Silt Fence Installation)
Coir Logs Step-by-Step Install coir log during periods 1 Dig a trench 2/3 the of dry riverbed or isolate area diameter of coir log (See silt fence installation). Secure Bank log with wooden or live stakes woven through coir log mesh and driven into earth. Stake log into Riverbed place every foot on both sides. Tie adjacent logs together with biodegradable twine. Compact soil around logs. Secure the upstream and downstream 2 ends by positioning coir logs so Dry coir they transition smoothly into a Stake driven log Bank stabilized bank. through log mesh 1/3 Riverbed 2/3 Alternatives using coir logs for securing toe of slope 3 depending upon site: a Crisscross layers of dormant cuttings 15 stems per linear foot (See live siltation). Trim willow after installation. c Spruce Tree Revetment Cabled trees with branches Bank Bank Coir Riverbed log b Brush Layering Riverbed Veg Mat Coir Soil/Topsoil log Coir log Earth Anchor Riverbed Coir Logs Coir logs are constructed of interwoven coconut fibers that are bound together with biodegradable netting. Commercially produced coir logs come in various lengths and diameters. The product needs to be selected specifically for the site. Fiber logs composed of other sturdy biodegradable materials may function equally as well. Applications for coir logs occur in many streambank, wetland and upland environments. The log provides temporary physical protection to a site while vegetation becomes established and biological protection takes over. The logs can provide a substrate for plant growth once the log decay process starts and protects native and newly installed plants growing adjacent to the log. -

Tensile Properties of Bamboo, Jute and Kenaf Mat-Reinforced Composite
Available online at www.sciencedirect.com ScienceDirect Energy Procedia 56 ( 2014 ) 72 – 79 11th Eco-Energy and Materials Science and Engineering (11th EMSES) Tensile Properties of Bamboo, Jute and Kenaf Mat-Reinforced Composite Toshihiko HOJOa,Zhilan XUb, Yuqiu YANGb*, Hiroyuki HAMADAa aKyoto Institute of Technology,Matsugasaki,Sakyo-ku, Kyoto, 6068585,Japan b Donghua University,Songjiang District,Shanghai, 201620,China Abstract Natural fibers, characterized by sustainability, have gained a considerable attention in recent years, due to their advantages of environmental acceptability and commercial viability. In this paper, several kinds of composites with natural fiber mat as reinforcement and unsaturated polyester(UP) as matrix, including jute/UP, kenaf/UP and bamboo/UP, were fabricated by hand lay-up and compression molding methods. Their tensile properties were tested and discussed, as well as the low cycle fatigue(LCF) behavior of three composites, which was compared with glass/UP. After the test, the fracture cross sectional observations were carried out on the selected test specimens using a scanning electron microscope(SEM),with a focus on the fracture morphologies. © 2014 Elsevier The Authors. Ltd. This Published is an open by access Elsevier article Ltd. under the CC BY-NC-ND license Peer-review(http://creativecommons.org/licenses/by-nc-nd/3.0/ under responsibility of COE of Sustainalble). Energy System, Rajamangala University of Technology Thanyaburi (RMUTT).Peer-review under responsibility of COE of Sustainalble Energy System, Rajamangala University of Technology Thanyaburi (RMUTT) Keywords: tensile property ; natural fiber mat; composites 1. Introduction Over the past few decades, there has been a growing interest in the use of natural fibers [1]. -

Mechanical Behaviour of Hybrid Composites Prepared Using Sisal-Pineapple-Kenaf Fibre
International Journal of Recent Technology and Engineering (IJRTE) ISSN: 2277-3878, Volume-8 Issue-5, January 2020 Mechanical Behaviour of Hybrid Composites Prepared using Sisal-Pineapple-Kenaf Fibre D Tamilvendan, G Mari Prabu, S Sivaraman, A. R. Ravikumar durability, tensile strength, impact strength, rupture strengths, Abstract: Variety of application use fibre reinforced composites stiffness and fatigue characteristics. Due to these numerous because of their intrinsic properties in mechanical strength, superior properties, they are extensively used in the machine renewability and low production cost compared to conventional parts like drive shafts, tanks, pressure vessels , automotive, materials. Natural fibres are environmentally friendly their use will not break the budget when used as an alternative to the combustion engines, thermal management, railway coaches regular materials. Reinforcement used in polymer is either and aircraft structures and power plant structures. man-made or natural. Man-made synthetic, metallic, Composites are made up of chemically distinct multiphase semi-synthetic, polymer fibres have superior specific strength but materials separated by distinct interface that exhibit better their high cost of production limits its application and feasibility to combination of properties compared to the constituent make composites. Recently there is a rise in use of natural fibres materials. Composite material is a combination of robust from various natural resources which are available abundantly. Composites based on natural fibres have their advantages of cost load-carrying material (known as reinforcement) imbedded in making the fibres from different vegetables, wood, animals and with weaker materials (known as matrix) differing in minerals. In this work a thorough and systematic inquiry composition on a macro scale.