Thermodynamic Properties of Multicomponent Mixtures from the Solution of Groups Approach to Direct Correlation Function Solution Theory
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

2D Potts Model Correlation Lengths
KOMA Revised version May D Potts Mo del Correlation Lengths Numerical Evidence for = at o d t Wolfhard Janke and Stefan Kappler Institut f ur Physik Johannes GutenbergUniversitat Mainz Staudinger Weg Mainz Germany Abstract We have studied spinspin correlation functions in the ordered phase of the twodimensional q state Potts mo del with q and at the rstorder transition p oint Through extensive Monte t Carlo simulations we obtain strong numerical evidence that the cor relation length in the ordered phase agrees with the exactly known and recently numerically conrmed correlation length in the disordered phase As a byproduct we nd the energy moments o t t d in the ordered phase at in very go o d agreement with a recent large t q expansion PACS numbers q Hk Cn Ha hep-lat/9509056 16 Sep 1995 Introduction Firstorder phase transitions have b een the sub ject of increasing interest in recent years They play an imp ortant role in many elds of physics as is witnessed by such diverse phenomena as ordinary melting the quark decon nement transition or various stages in the evolution of the early universe Even though there exists already a vast literature on this sub ject many prop erties of rstorder phase transitions still remain to b e investigated in detail Examples are nitesize scaling FSS the shap e of energy or magnetization distributions partition function zeros etc which are all closely interrelated An imp ortant approach to attack these problems are computer simulations Here the available system sizes are necessarily -

Full and Unbiased Solution of the Dyson-Schwinger Equation in the Functional Integro-Differential Representation
PHYSICAL REVIEW B 98, 195104 (2018) Full and unbiased solution of the Dyson-Schwinger equation in the functional integro-differential representation Tobias Pfeffer and Lode Pollet Department of Physics, Arnold Sommerfeld Center for Theoretical Physics, University of Munich, Theresienstrasse 37, 80333 Munich, Germany (Received 17 March 2018; revised manuscript received 11 October 2018; published 2 November 2018) We provide a full and unbiased solution to the Dyson-Schwinger equation illustrated for φ4 theory in 2D. It is based on an exact treatment of the functional derivative ∂/∂G of the four-point vertex function with respect to the two-point correlation function G within the framework of the homotopy analysis method (HAM) and the Monte Carlo sampling of rooted tree diagrams. The resulting series solution in deformations can be considered as an asymptotic series around G = 0 in a HAM control parameter c0G, or even a convergent one up to the phase transition point if shifts in G can be performed (such as by summing up all ladder diagrams). These considerations are equally applicable to fermionic quantum field theories and offer a fresh approach to solving functional integro-differential equations beyond any truncation scheme. DOI: 10.1103/PhysRevB.98.195104 I. INTRODUCTION was not solved and differences with the full, exact answer could be seen when the correlation length increases. Despite decades of research there continues to be a need In this work, we solve the full DSE by writing them as a for developing novel methods for strongly correlated systems. closed set of integro-differential equations. Within the HAM The standard Monte Carlo approaches [1–5] are convergent theory there exists a semianalytic way to treat the functional but suffer from a prohibitive sign problem, scaling exponen- derivatives without resorting to an infinite expansion of the tially in the system volume [6]. -

Notes on Statistical Field Theory
Lecture Notes on Statistical Field Theory Kevin Zhou [email protected] These notes cover statistical field theory and the renormalization group. The primary sources were: • Kardar, Statistical Physics of Fields. A concise and logically tight presentation of the subject, with good problems. Possibly a bit too terse unless paired with the 8.334 video lectures. • David Tong's Statistical Field Theory lecture notes. A readable, easygoing introduction covering the core material of Kardar's book, written to seamlessly pair with a standard course in quantum field theory. • Goldenfeld, Lectures on Phase Transitions and the Renormalization Group. Covers similar material to Kardar's book with a conversational tone, focusing on the conceptual basis for phase transitions and motivation for the renormalization group. The notes are structured around the MIT course based on Kardar's textbook, and were revised to include material from Part III Statistical Field Theory as lectured in 2017. Sections containing this additional material are marked with stars. The most recent version is here; please report any errors found to [email protected]. 2 Contents Contents 1 Introduction 3 1.1 Phonons...........................................3 1.2 Phase Transitions......................................6 1.3 Critical Behavior......................................8 2 Landau Theory 12 2.1 Landau{Ginzburg Hamiltonian.............................. 12 2.2 Mean Field Theory..................................... 13 2.3 Symmetry Breaking.................................... 16 3 Fluctuations 19 3.1 Scattering and Fluctuations................................ 19 3.2 Position Space Fluctuations................................ 20 3.3 Saddle Point Fluctuations................................. 23 3.4 ∗ Path Integral Methods.................................. 24 4 The Scaling Hypothesis 29 4.1 The Homogeneity Assumption............................... 29 4.2 Correlation Lengths.................................... 30 4.3 Renormalization Group (Conceptual).......................... -

10 Basic Aspects of CFT
10 Basic aspects of CFT An important break-through occurred in 1984 when Belavin, Polyakov and Zamolodchikov [BPZ84] applied ideas of conformal invariance to classify the possible types of critical behaviour in two dimensions. These ideas had emerged earlier in string theory and mathematics, and in fact go backto earlier (1970) work of Polyakov [Po70] in which global conformal invariance is used to constrain the form of correlation functions in d-dimensional the- ories. It is however only by imposing local conformal invariance in d =2 that this approach becomes really powerful. In particular, it immediately permitted a full classification of an infinite family of conformally invariant theories (the so-called “minimal models”) having a finite number of funda- mental (“primary”) fields, and the exact computation of the corresponding critical exponents. In the aftermath of these developments, conformal field theory (CFT) became for some years one of the most hectic research fields of theoretical physics, and indeed has remained a very active area up to this date. This chapter focusses on the basic aspects of CFT, with a special emphasis on the ingredients which will allow us to tackle the geometrically defined loop models via the so-called Coulomb Gas (CG) approach. The CG technique will be exposed in the following chapter. The aim is to make the presentation self- contained while remaining rather brief; the reader interested in more details should turn to the comprehensive textbook [DMS87] or the Les Houches volume [LH89]. 10.1 Global conformal invariance A conformal transformation in d dimensions is an invertible mapping x x′ → which multiplies the metric tensor gµν (x) by a space-dependent scale factor: gµ′ ν (x′)=Λ(x)gµν (x). -

Introduction to Two-Dimensional Conformal Field Theory
Introduction to two-dimensional conformal field theory Sylvain Ribault CEA Saclay, Institut de Physique Th´eorique [email protected] February 5, 2019 Abstract We introduce conformal field theory in two dimensions, from the basic principles to some of the simplest models. From the representations of the Virasoro algebra on the one hand, and the state-field correspondence on the other hand, we deduce Ward identities and Belavin{Polyakov{Zamolodchikov equations for correlation functions. We then explain the principles of the conformal bootstrap method, and introduce conformal blocks. This allows us to define and solve minimal models and Liouville theory. We also introduce the free boson with its abelian affine Lie algebra. Lecture notes for the \Young Researchers Integrability School", Wien 2019, based on the earlier lecture notes [1]. Estimated length: 7 lectures of 45 minutes each. Material that may be skipped in the lectures is in green boxes. Exercises are in green boxes when their statements are not part of the lectures' text; this does not make them less interesting as exercises. 1 Contents 0 Introduction2 1 The Virasoro algebra and its representations3 1.1 Algebra.....................................3 1.2 Representations.................................4 1.3 Null vectors and degenerate representations.................5 2 Conformal field theory6 2.1 Fields......................................6 2.2 Correlation functions and Ward identities...................8 2.3 Belavin{Polyakov{Zamolodchikov equations................. 10 2.4 Free boson.................................... 10 3 Conformal bootstrap 12 3.1 Single-valuedness................................ 12 3.2 Operator product expansion and crossing symmetry............. 13 3.3 Degenerate fields and the fusion product................... 16 4 Minimal models 17 4.1 Diagonal minimal models........................... -
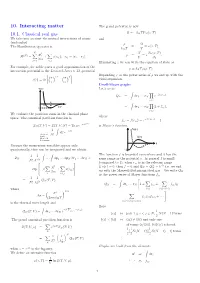
10. Interacting Matter 10.1. Classical Real
10. Interacting matter The grand potential is now Ω= k TVω(z,T ) 10.1. Classical real gas − B We take into account the mutual interactions of atoms and (molecules) p Ω The Hamiltonian operator is = = ω(z,T ) kBT −V N p2 N ∂ω(z,T ) H(N) = i + v(r ), r = r r . ρ = = z . 2m ij ij | i − j | V ∂z i=1 i<j X X Eliminating z we can write the equation of state as For example, for noble gases a good approximation of the p = k T ϕ(ρ,T ). interaction potential is the Lennard-Jones 6–12 -potential B Expanding ϕ as the power series of ρ we end up with the σ 12 σ 6 v(r)=4ǫ . virial expansion. r − r Ursell-Mayer graphs V ( r ) Let’s write Q = dr r e−βv(rij ) N 1 · · · N i<j Z Y r 0 s r 6 = dr r (1 + f ), e 1 / r 1 · · · N ij Z i<j We evaluate the partition sums in the classical phase Y where space. The canonical partition function is f = f(r )= e−βv(rij ) 1 ij ij − −βH(N) ZN (T, V )= Z(T,V,N) = Tr N e is Mayer’s function. 1 V ( r ) dΓ e−βH . classical−→ N! limit, Maxwell- Z f ( r ) Boltzman Because the momentum variables appear only r quadratically, they can be integrated and we obtain - 1 1 1 The function f is bounded everywhere and it has the ZN = dp1 dpN dr1 drN same range as the potential v. -

Recursive Graphical Solution of Closed Schwinger-Dyson
Recursive Graphical Solution of Closed Schwinger-Dyson Equations in φ4-Theory – Part1: Generation of Connected and One-Particle Irreducible Feynman Diagrams Axel Pelster and Konstantin Glaum Institut f¨ur Theoretische Physik, Freie Universit¨at Berlin, Arnimallee 14, 14195 Berlin, Germany [email protected],[email protected] (Dated: November 11, 2018) Using functional derivatives with respect to the free correlation function we derive a closed set of Schwinger-Dyson equations in φ4-theory. Its conversion to graphical recursion relations allows us to systematically generate all connected and one-particle irreducible Feynman diagrams for the two- and four-point function together with their weights. PACS numbers: 05.70.Fh,64.60.-i I. INTRODUCTION Quantum and statistical field theory investigate the influence of field fluctuations on the n-point functions. Interactions lead to an infinite hierarchy of Schwinger-Dyson equations for the n-point functions [1–6]. These integral equations can only be closed approximately, for instance, by the well-established the self-consistent method of Kadanoff and Baym [7]. Recently, it has been shown that the Schwinger-Dyson equations of QED can be closed in a certain functional- analytic sense [8]. Using functional derivatives with respect to the free propagators and the interaction [8–14] two closed sets of equations were derived. The first one involves the connected electron and two-point function as well as the connected three-point function, whereas the second one determines the electron and photon self-energy as well as the one-particle irreducible three-point function. Their conversion to graphical recursion relations leads to a systematic graphical generation of all connected and one-particle irreducible Feynman diagrams in QED, respectively. -

Dirty Tricks for Statistical Mechanics
Dirty tricks for statistical mechanics Martin Grant Physics Department, McGill University c MG, August 2004, version 0.91 ° ii Preface These are lecture notes for PHYS 559, Advanced Statistical Mechanics, which I’ve taught at McGill for many years. I’m intending to tidy this up into a book, or rather the first half of a book. This half is on equilibrium, the second half would be on dynamics. These were handwritten notes which were heroically typed by Ryan Porter over the summer of 2004, and may someday be properly proof-read by me. Even better, maybe someday I will revise the reasoning in some of the sections. Some of it can be argued better, but it was too much trouble to rewrite the handwritten notes. I am also trying to come up with a good book title, amongst other things. The two titles I am thinking of are “Dirty tricks for statistical mechanics”, and “Valhalla, we are coming!”. Clearly, more thinking is necessary, and suggestions are welcome. While these lecture notes have gotten longer and longer until they are al- most self-sufficient, it is always nice to have real books to look over. My favorite modern text is “Lectures on Phase Transitions and the Renormalisation Group”, by Nigel Goldenfeld (Addison-Wesley, Reading Mass., 1992). This is referred to several times in the notes. Other nice texts are “Statistical Physics”, by L. D. Landau and E. M. Lifshitz (Addison-Wesley, Reading Mass., 1970) par- ticularly Chaps. 1, 12, and 14; “Statistical Mechanics”, by S.-K. Ma (World Science, Phila., 1986) particularly Chaps. -

Some Aspects of Conformal Field Theories on the Plane and Higher Genus Riemann Surfaces
Pramg.na - J. Phys., Vol. 35, No. 3, September 1990, pp. 205-286. © Printed in India. Some aspects of conformal field theories on the plane and higher genus Riemann surfaces ASHOKE SEN Tata Institute of Fundamental Research, Homi Bhabha Road, Bombay 400005, India MS received 6 June 1990 Al~traet. We review some aspects of conformal field theories on the plane as well as on higher genus Riemann surfaces. Keywords. Conformal field theory; Riemann surfaces. PACS No. 11.10 1. Introduction Conformally invariant two dimensional field theories (CFT) have become the subject of intense investigation in recent years. In this article I shall try to give a general introduction' to conformal field theory with special emphasis on a particular class of conformal field theories, known as rational conformal field theories (RCFT). I shall begin by discussing the reasons for the recent upsurge of interest in these theories, and then discuss the various properties of these theories in some detail. One of the two main applications of two dimensional conformal field theories is that they describe the critical behavior of many known two dimensional statistical mechanical models. In order to understand this connection we must first understand the meaning of conformal invariance. In any dimension, conformal invariance refers to a group of coordinate transformations which leave the angle between any two intersecting lines fixed. Obviously the Poincare group, consisting of translations and rotations have this property, and hence they form part of the conformal group. Another transformation which has this property is the scale transformation. In general the conformal group has other elements also which we shall discuss later, but for the purpose of understanding the connection to the critical behavior of statistical models, the above properties are enough. -

Statistical Field Theory University of Cambridge Part III Mathematical Tripos
Preprint typeset in JHEP style - HYPER VERSION Michaelmas Term, 2017 Statistical Field Theory University of Cambridge Part III Mathematical Tripos David Tong Department of Applied Mathematics and Theoretical Physics, Centre for Mathematical Sciences, Wilberforce Road, Cambridge, CB3 OBA, UK http://www.damtp.cam.ac.uk/user/tong/sft.html [email protected] { 1 { Recommended Books and Resources There are a large number of books which cover the material in these lectures, although often from very different perspectives. They have titles like \Critical Phenomena", \Phase Transitions", \Renormalisation Group" or, less helpfully, \Advanced Statistical Mechanics". Here are some that I particularly like • Nigel Goldenfeld, Phase Transitions and the Renormalization Group A great book, covering the basic material that we'll need and delving deeper in places. • Mehran Kardar, Statistical Physics of Fields The second of two volumes on statistical mechanics. It cuts a concise path through the subject, at the expense of being a little telegraphic in places. It is based on lecture notes which you can find on the web; a link is given on the course website. • John Cardy, Scaling and Renormalisation in Statistical Physics A beautiful little book from one of the masters of conformal field theory. It covers the material from a slightly different perspective than these lectures, with more focus on renormalisation in real space. • Chaikin and Lubensky, Principles of Condensed Matter Physics • Shankar, Quantum Field Theory and Condensed Matter Both of these are more all-round condensed matter books, but with substantial sections on critical phenomena and the renormalisation group. Chaikin and Lubensky is more traditional, and packed full of content. -
![Arxiv:0704.3906V2 [Quant-Ph]](https://docslib.b-cdn.net/cover/6018/arxiv-0704-3906v2-quant-ph-2066018.webp)
Arxiv:0704.3906V2 [Quant-Ph]
Area laws in quantum systems: mutual information and correlations Michael M. Wolf1, Frank Verstraete2, Matthew B. Hastings3, J. Ignacio Cirac1 1 Max-Planck-Institut f¨ur Quantenoptik, Hans-Kopfermann-Str.1, 85748 Garching, Germany. 2 Fakult¨at f¨ur Physik, Universit¨at Wien, Boltzmanngasse 5, A-1090 Wien, Austria. 3 Center for Non-linear Studies and Theoretical Division, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA (Dated: March 10, 2008) The holographic principle states that on a fundamental level the information content of a region should depend on its surface area rather than on its volume. In this paper we show that this phenomenon not only emerges in the search for new Planck-scale laws but also in lattice models of classical and quantum physics: the information contained in part of a system in thermal equilibrium obeys an area law. While the maximal information per unit area depends classically only on the number of degrees of freedom, it may diverge as the inverse temperature in quantum systems. It is shown that an area law is generally implied by a finite correlation length when measured in terms of the mutual information. Correlations are information of one system about another. The study of correlations in equilibrium lattice models comes in two flavors. The more traditional approach is the investiga- tion of the decay of two-point correlations with the distance. A lot of knowledge has been acquired in Condensed Matter Physics in this direction and is now being used and developed A further in the study of entanglement in Quantum Information Theory [1, 2, 3]. -

4. Introducing Conformal Field Theory
4. Introducing Conformal Field Theory The purpose of this section is to get comfortable with the basic language of two dimen- sional conformal field theory4.Thisisatopicwhichhasmanyapplicationsoutsideof string theory, most notably in statistical physics where it o↵ers a description of critical phenomena. Moreover, it turns out that conformal field theories in two dimensions provide rare examples of interacting, yet exactly solvable, quantum field theories. In recent years, attention has focussed on conformal field theories in higher dimensions due to their role in the AdS/CFT correspondence. A conformal transformation is a change of coordinates σ↵ σ˜↵(σ)suchthatthe ! metric changes by g (σ) ⌦2(σ)g (σ)(4.1) ↵ ! ↵ A conformal field theory (CFT) is a field theory which is invariant under these transfor- mations. This means that the physics of the theory looks the same at all length scales. Conformal field theories care about angles, but not about distances. Atransformationoftheform(4.1)hasadi↵erentinterpretationdependingonwhether we are considering a fixed background metric g↵, or a dynamical background metric. When the metric is dynamical, the transformation is a di↵eomorphism; this is a gauge symmetry. When the background is fixed, the transformation should be thought of as an honest, physical symmetry, taking the point σ↵ to pointσ ˜↵. This is now a global symmetry with the corresponding conserved currents. In the context of string theory in the Polyakov formalism, the metric is dynamical and the transformations (4.1) are residual gauge transformations: di↵eomorphisms which can be undone by a Weyl transformation. In contrast, in this section we will be primarily interested in theories defined on fixed backgrounds.