Chimericseq Quick User Guide for Windows Fwu-Shan Shieh Version 4.1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

At—At, Batch—Execute Commands at a Later Time
at—at, batch—execute commands at a later time at [–csm] [–f script] [–qqueue] time [date] [+ increment] at –l [ job...] at –r job... batch at and batch read commands from standard input to be executed at a later time. at allows you to specify when the commands should be executed, while jobs queued with batch will execute when system load level permits. Executes commands read from stdin or a file at some later time. Unless redirected, the output is mailed to the user. Example A.1 1 at 6:30am Dec 12 < program 2 at noon tomorrow < program 3 at 1945 pm August 9 < program 4 at now + 3 hours < program 5 at 8:30am Jan 4 < program 6 at -r 83883555320.a EXPLANATION 1. At 6:30 in the morning on December 12th, start the job. 2. At noon tomorrow start the job. 3. At 7:45 in the evening on August 9th, start the job. 4. In three hours start the job. 5. At 8:30 in the morning of January 4th, start the job. 6. Removes previously scheduled job 83883555320.a. awk—pattern scanning and processing language awk [ –fprogram–file ] [ –Fc ] [ prog ] [ parameters ] [ filename...] awk scans each input filename for lines that match any of a set of patterns specified in prog. Example A.2 1 awk '{print $1, $2}' file 2 awk '/John/{print $3, $4}' file 3 awk -F: '{print $3}' /etc/passwd 4 date | awk '{print $6}' EXPLANATION 1. Prints the first two fields of file where fields are separated by whitespace. 2. Prints fields 3 and 4 if the pattern John is found. -

Unix Introduction
Unix introduction Mikhail Dozmorov Summer 2018 Mikhail Dozmorov Unix introduction Summer 2018 1 / 37 What is Unix Unix is a family of operating systems and environments that exploits the power of linguistic abstractions to perform tasks Unix is not an acronym; it is a pun on “Multics”. Multics was a large multi-user operating system that was being developed at Bell Labs shortly before Unix was created in the early ’70s. Brian Kernighan is credited with the name. All computational genomics is done in Unix http://www.read.seas.harvard.edu/~kohler/class/aosref/ritchie84evolution.pdfMikhail Dozmorov Unix introduction Summer 2018 2 / 37 History of Unix Initial file system, command interpreter (shell), and process management started by Ken Thompson File system and further development from Dennis Ritchie, as well as Doug McIlroy and Joe Ossanna Vast array of simple, dependable tools that each do one simple task Ken Thompson (sitting) and Dennis Ritchie working together at a PDP-11 Mikhail Dozmorov Unix introduction Summer 2018 3 / 37 Philosophy of Unix Vast array of simple, dependable tools Each do one simple task, and do it really well By combining these tools, one can conduct rather sophisticated analyses The Linux help philosophy: “RTFM” (Read the Fine Manual) Mikhail Dozmorov Unix introduction Summer 2018 4 / 37 Know your Unix Unix users spend a lot of time at the command line In Unix, a word is worth a thousand mouse clicks Mikhail Dozmorov Unix introduction Summer 2018 5 / 37 Unix systems Three common types of laptop/desktop operating systems: Windows, Mac, Linux. Mac and Linux are both Unix-like! What that means for us: Unix-like operating systems are equipped with “shells”" that provide a command line user interface. -

Introduction to Unix
Introduction to Unix Rob Funk <[email protected]> University Technology Services Workstation Support http://wks.uts.ohio-state.edu/ University Technology Services Course Objectives • basic background in Unix structure • knowledge of getting started • directory navigation and control • file maintenance and display commands • shells • Unix features • text processing University Technology Services Course Objectives Useful commands • working with files • system resources • printing • vi editor University Technology Services In the Introduction to UNIX document 3 • shell programming • Unix command summary tables • short Unix bibliography (also see web site) We will not, however, be covering these topics in the lecture. Numbers on slides indicate page number in book. University Technology Services History of Unix 7–8 1960s multics project (MIT, GE, AT&T) 1970s AT&T Bell Labs 1970s/80s UC Berkeley 1980s DOS imitated many Unix ideas Commercial Unix fragmentation GNU Project 1990s Linux now Unix is widespread and available from many sources, both free and commercial University Technology Services Unix Systems 7–8 SunOS/Solaris Sun Microsystems Digital Unix (Tru64) Digital/Compaq HP-UX Hewlett Packard Irix SGI UNICOS Cray NetBSD, FreeBSD UC Berkeley / the Net Linux Linus Torvalds / the Net University Technology Services Unix Philosophy • Multiuser / Multitasking • Toolbox approach • Flexibility / Freedom • Conciseness • Everything is a file • File system has places, processes have life • Designed by programmers for programmers University Technology Services -

Praat Scripting Tutorial
Praat Scripting Tutorial Eleanor Chodroff Newcastle University July 2019 Praat Acoustic analysis program Best known for its ability to: Visualize, label, and segment audio files Perform spectral and temporal analyses Synthesize and manipulate speech Praat Scripting Praat: not only a program, but also a language Why do I want to know Praat the language? AUTOMATE ALL THE THINGS Praat Scripting Why can’t I just modify others’ scripts? Honestly: power, flexibility, control Insert: all the gifs of ‘you can do it’ and ‘you got this’ and thumbs up Praat Scripting Goals ~*~Script first for yourself, then for others~*~ • Write Praat scripts quickly, effectively, and “from scratch” • Learn syntax and structure of the language • Handle various input/output combinations Tutorial Overview 1) Praat: Big Picture 2) Getting started 3) Basic syntax 4) Script types + Practice • Wav files • Measurements • TextGrids • Other? Praat: Big Picture 1) Similar to other languages you may (or may not) have used before • String and numeric variables • For-loops, if else statements, while loops • Regular expression matching • Interpreted language (not compiled) Praat: Big Picture 2) Almost everything is a mouse click! i.e., Praat is a GUI scripting language GUI = Graphical User Interface, i.e., the Objects window If you ever get lost while writing a Praat script, click through the steps using the GUI Getting Started Open a Praat script From the toolbar, select Praat à New Praat script Save immediately! Save frequently! Script Goals and Input/Output • Consider what -

Deviceinstaller User Guide
Device Installer User Guide Part Number 900-325 Revision C 03/18 Table of Contents 1. Overview ...................................................................................................................................... 1 2. Devices ........................................................................................................................................ 2 Choose the Network Adapter for Communication ....................................................................... 2 Search for All Devices on the Network ........................................................................................ 2 Change Views .............................................................................................................................. 2 Add a Device to the List ............................................................................................................... 3 View Device Details ..................................................................................................................... 3 Device Lists ................................................................................................................................. 3 Save the Device List ................................................................................................................ 3 Open the Device List ............................................................................................................... 4 Print the Device List ................................................................................................................ -

Sys, Shutil, and OS Modules
This video will introduce the sys, shutil, glob, and os modules. 1 The sys module contains a few tools that are often useful and will be used in this course. The first tool is the argv function which allows the script to request a parameter when the script is run. We will use this function in a few weeks when importing scripts into ArcGIS. If a zero is specified for the argv function, then it will return the location of the script on the computer. This can be helpful if the script needs to look for a supplemental file, such as a template, that is located in the same folder as the script. The exit function stops the script when it reach the exit statement. The function can be used to stop a script when a certain condition occurs – such as when a user specifies an invalid parameter. This allows the script to stop gracefully rather than crashing. The exit function is also useful when troubleshooting a script. 2 The shutil module contains tools for moving and deleting files. The copy tool makes a copy of a file in a new location. The copytree tool makes a copy of an entire folder in a new location. The rmtree tool deletes an entire folder. Note that this tool will not send items to the recycling bin and it will not ask for confirmation. The tool will fail if it encounters any file locks in the folder. 3 The os module contains operating system functions – many of which are often useful in a script. -

ANSWERS ΤΟ EVEN-Numbered
8 Answers to Even-numbered Exercises 2.1. WhatExplain the following unexpected are result: two ways you can execute a shell script when you do not have execute permission for the file containing the script? Can you execute a shell script if you do not have read permission for the file containing the script? You can give the name of the file containing the script as an argument to the shell (for example, bash scriptfile or tcsh scriptfile, where scriptfile is the name of the file containing the script). Under bash you can give the following command: $ . scriptfile Under both bash and tcsh you can use this command: $ source scriptfile Because the shell must read the commands from the file containing a shell script before it can execute the commands, you must have read permission for the file to execute a shell script. 4.3. AssumeWhat is the purpose ble? you have made the following assignment: $ person=zach Give the output of each of the following commands. a. echo $person zach b. echo '$person' $person c. echo "$person" zach 1 2 6.5. Assumengs. the /home/zach/grants/biblios and /home/zach/biblios directories exist. Specify Zach’s working directory after he executes each sequence of commands. Explain what happens in each case. a. $ pwd /home/zach/grants $ CDPATH=$(pwd) $ cd $ cd biblios After executing the preceding commands, Zach’s working directory is /home/zach/grants/biblios. When CDPATH is set and the working directory is not specified in CDPATH, cd searches the working directory only after it searches the directories specified by CDPATH. -

Linux Networking Cookbook.Pdf
Linux Networking Cookbook ™ Carla Schroder Beijing • Cambridge • Farnham • Köln • Paris • Sebastopol • Taipei • Tokyo Linux Networking Cookbook™ by Carla Schroder Copyright © 2008 O’Reilly Media, Inc. All rights reserved. Printed in the United States of America. Published by O’Reilly Media, Inc., 1005 Gravenstein Highway North, Sebastopol, CA 95472. O’Reilly books may be purchased for educational, business, or sales promotional use. Online editions are also available for most titles (safari.oreilly.com). For more information, contact our corporate/institutional sales department: (800) 998-9938 or [email protected]. Editor: Mike Loukides Indexer: John Bickelhaupt Production Editor: Sumita Mukherji Cover Designer: Karen Montgomery Copyeditor: Derek Di Matteo Interior Designer: David Futato Proofreader: Sumita Mukherji Illustrator: Jessamyn Read Printing History: November 2007: First Edition. Nutshell Handbook, the Nutshell Handbook logo, and the O’Reilly logo are registered trademarks of O’Reilly Media, Inc. The Cookbook series designations, Linux Networking Cookbook, the image of a female blacksmith, and related trade dress are trademarks of O’Reilly Media, Inc. Java™ is a trademark of Sun Microsystems, Inc. .NET is a registered trademark of Microsoft Corporation. Many of the designations used by manufacturers and sellers to distinguish their products are claimed as trademarks. Where those designations appear in this book, and O’Reilly Media, Inc. was aware of a trademark claim, the designations have been printed in caps or initial caps. While every precaution has been taken in the preparation of this book, the publisher and author assume no responsibility for errors or omissions, or for damages resulting from the use of the information contained herein. -

Sample Applications User Guide Release 16.04.0
Sample Applications User Guide Release 16.04.0 April 12, 2016 CONTENTS 1 Introduction 1 1.1 Documentation Roadmap...............................1 2 Command Line Sample Application2 2.1 Overview........................................2 2.2 Compiling the Application...............................2 2.3 Running the Application................................3 2.4 Explanation.......................................3 3 Ethtool Sample Application5 3.1 Compiling the Application...............................5 3.2 Running the Application................................5 3.3 Using the application.................................5 3.4 Explanation.......................................6 3.5 Ethtool interface....................................6 4 Exception Path Sample Application8 4.1 Overview........................................8 4.2 Compiling the Application...............................9 4.3 Running the Application................................9 4.4 Explanation....................................... 10 5 Hello World Sample Application 13 5.1 Compiling the Application............................... 13 5.2 Running the Application................................ 13 5.3 Explanation....................................... 13 6 Basic Forwarding Sample Application 15 6.1 Compiling the Application............................... 15 6.2 Running the Application................................ 15 6.3 Explanation....................................... 15 7 RX/TX Callbacks Sample Application 20 7.1 Compiling the Application.............................. -
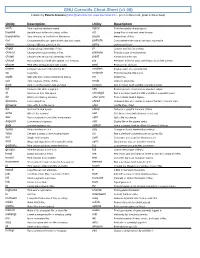
GNU Coreutils Cheat Sheet (V1.00) Created by Peteris Krumins ([email protected], -- Good Coders Code, Great Coders Reuse)
GNU Coreutils Cheat Sheet (v1.00) Created by Peteris Krumins ([email protected], www.catonmat.net -- good coders code, great coders reuse) Utility Description Utility Description arch Print machine hardware name nproc Print the number of processors base64 Base64 encode/decode strings or files od Dump files in octal and other formats basename Strip directory and suffix from file names paste Merge lines of files cat Concatenate files and print on the standard output pathchk Check whether file names are valid or portable chcon Change SELinux context of file pinky Lightweight finger chgrp Change group ownership of files pr Convert text files for printing chmod Change permission modes of files printenv Print all or part of environment chown Change user and group ownership of files printf Format and print data chroot Run command or shell with special root directory ptx Permuted index for GNU, with keywords in their context cksum Print CRC checksum and byte counts pwd Print current directory comm Compare two sorted files line by line readlink Display value of a symbolic link cp Copy files realpath Print the resolved file name csplit Split a file into context-determined pieces rm Delete files cut Remove parts of lines of files rmdir Remove directories date Print or set the system date and time runcon Run command with specified security context dd Convert a file while copying it seq Print sequence of numbers to standard output df Summarize free disk space setuidgid Run a command with the UID and GID of a specified user dir Briefly list directory -

Bedtools Documentation Release 2.30.0
Bedtools Documentation Release 2.30.0 Quinlan lab @ Univ. of Utah Jan 23, 2021 Contents 1 Tutorial 3 2 Important notes 5 3 Interesting Usage Examples 7 4 Table of contents 9 5 Performance 169 6 Brief example 173 7 License 175 8 Acknowledgments 177 9 Mailing list 179 i ii Bedtools Documentation, Release 2.30.0 Collectively, the bedtools utilities are a swiss-army knife of tools for a wide-range of genomics analysis tasks. The most widely-used tools enable genome arithmetic: that is, set theory on the genome. For example, bedtools allows one to intersect, merge, count, complement, and shuffle genomic intervals from multiple files in widely-used genomic file formats such as BAM, BED, GFF/GTF, VCF. While each individual tool is designed to do a relatively simple task (e.g., intersect two interval files), quite sophisticated analyses can be conducted by combining multiple bedtools operations on the UNIX command line. bedtools is developed in the Quinlan laboratory at the University of Utah and benefits from fantastic contributions made by scientists worldwide. Contents 1 Bedtools Documentation, Release 2.30.0 2 Contents CHAPTER 1 Tutorial We have developed a fairly comprehensive tutorial that demonstrates both the basics, as well as some more advanced examples of how bedtools can help you in your research. Please have a look. 3 Bedtools Documentation, Release 2.30.0 4 Chapter 1. Tutorial CHAPTER 2 Important notes • As of version 2.28.0, bedtools now supports the CRAM format via the use of htslib. Specify the reference genome associated with your CRAM file via the CRAM_REFERENCE environment variable. -

07 07 Unixintropart2 Lucio Week 3
Unix Basics Command line tools Daniel Lucio Overview • Where to use it? • Command syntax • What are commands? • Where to get help? • Standard streams(stdin, stdout, stderr) • Pipelines (Power of combining commands) • Redirection • More Information Introduction to Unix Where to use it? • Login to a Unix system like ’kraken’ or any other NICS/ UT/XSEDE resource. • Download and boot from a Linux LiveCD either from a CD/DVD or USB drive. • http://www.puppylinux.com/ • http://www.knopper.net/knoppix/index-en.html • http://www.ubuntu.com/ Introduction to Unix Where to use it? • Install Cygwin: a collection of tools which provide a Linux look and feel environment for Windows. • http://cygwin.com/index.html • https://newton.utk.edu/bin/view/Main/Workshop0InstallingCygwin • Online terminal emulator • http://bellard.org/jslinux/ • http://cb.vu/ • http://simpleshell.com/ Introduction to Unix Command syntax $ command [<options>] [<file> | <argument> ...] Example: cp [-R [-H | -L | -P]] [-fi | -n] [-apvX] source_file target_file Introduction to Unix What are commands? • An executable program (date) • A command built into the shell itself (cd) • A shell program/function • An alias Introduction to Unix Bash commands (Linux) alias! crontab! false! if! mknod! ram! strace! unshar! apropos! csplit! fdformat! ifconfig! more! rcp! su! until! apt-get! cut! fdisk! ifdown! mount! read! sudo! uptime! aptitude! date! fg! ifup! mtools! readarray! sum! useradd! aspell! dc! fgrep! import! mtr! readonly! suspend! userdel! awk! dd! file! install! mv! reboot! symlink!