Jimmunol.1002449.Full.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Separation of Β-Receptor Blockers and Analogs by Capillary Liquid Chromatography
ORIGINAL ARTICLES College of Pharmaceutical Sciences, Zhejiang University, Hangzhou, China Separation of b-receptor blockers and analogs by Capillary Liquid Chromatography (CLC) and Pressurized Capillary Electrochromatography (pCEC) using a vancomycin chiral stationary phase column Zhongyi Chen, Su Zeng, Tongwei Yao Received September 15, 2006, accepted October 28, 2006 Tongwei Jao, Department of Pharmaceutical Analysis and Drug Metabolism, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou, 310031, China [email protected] Pharmazie 62: 585–592 (2007) doi: 10.1691/ph.2007.8.6194 Enantiomeric separation of chiral pharmaceuticals was carried out by means of in capillary liquid chroma- tography (CLC) and pressurized capillary electrochromatography (pCEC) using a vancomycin chiral sta- tionary phase (CSP). A 100 mm I.D. fused-silica capillary was packed with 5 mm diameter silica particles modified with vancomycin. Enantiomeric resolution of fifteen b-receptor blockers and analogs was stu- died by polar organic CLC mode and reversed-phase pCEC mode using mobile phases containing methanol-isopropanol-acetic acid-triethylamine and TEAA buffer-methanol, respectively. Several factors affecting chiral separation were investigated in both CLC and pCEC mode. Good enantiomeric resolution was achieved by CLC mode for propranolol, celiprolol, esmolol, bisoprolol, atenolol, metoprolol and car- teolol using methanol-isopropanol-acetic acid-triethylamine (70 : 30 : 0.05 : 0.05, v/v/v/v) as mobile phase and for clenbuterol, bambuterol, terbutaline, and salbutamol using methanol-isopropanol-acetic acid- triethylamine (50 : 50 : 0.05 : 005 or 50 : 50: 0.025 : 0.05, v/v/v/v) as mobile phase. The baseline was achieved by pCEC mode for the separation of esmolol, bisoprolol, atenolol, metoprolol, carteolol in the mobile phase containing MeOH-0.05%TEAA (pH 7.0) (90 : 10, v/v) (–10 kV), and that of propranolol and celiprolol in the mobile phase containing MeOH-0.025%TEAA (pH 7.0) (90 : 10, v/v)(–10 kV). -

Clenbuterol Human Effects the Effect of Clenbuterol in Humans Is Researched Through Examining the History and Regulations of the Drug
Clenbuterol Human Effects The effect of clenbuterol in humans is researched through examining the history and regulations of the drug. Specifically, Alberto Contador’s case is considered. Tag Words: Clenbuterol; drugs; Beta-2 Agonist; Effects; Thermogenic; Fat; Harmful; Authors: Jessie Yeh, Horace Lau, Danielle Lovisone with Julie M. Fagan, Ph.D. Summary (written by Danielle Lovisone) As a sympathomimetic and Beta-2 agonist, clenbuterol have several deleterious effects on the human body. The drug acts as a thermogenic stimulant, increasing lean muscle mass and respiratory efficiency while reducing fat. Cases on clenbuterol, including animal tests and human occurrences, support these unnatural and potentially harmful effects. With this, athletes and body builders have recently increased their use of the drug. Particularly, Alberto Contador has recently been targeted for having traces of clenbuterol in a urine drug test. Contador claims, instead of doping, this trace amount was unknowingly received from ingesting beef in Spain during the 2010 Tour de France. Although clenbuterol is banned in most areas of the world, this explanation seems plausible because the drug is poorly regulated by organizations such as the FDA. To examine Contador’s case further, our group compiled research on clenbuterol to ultimately hypothesize that Contador received this trace amount from contaminated beef. Our findings were submitted to the World Anti-Doping Agency as part of our Service Project. Video Link Class project 2010 fall: www.youtube.com/watch?v=ZTeJiBvbLhA The Issue: Clenbuterol The Effects of Clenbuterol on the Human Body By Jessie Yeh What is Clenbuterol? Clenbuterol is a chemical compound closely resembling the structure of an amine. -

Asthma Medications
Relievers / Rescue / Bronchodilators Asthma Short-acting Beta Agonists 2 Medications Ipratropium Bromide ProAir ProAir RespiClick Proventil Ventolin Xopenex albuterol sulfate albuterol sulfate dry powder albuterol sulfate albuterol sulfate levalbuterol tartrate 90mcg 90mcg 90mcg 90mcg 45mcg Teva Teva Merck GlaxoSmithKline Sunovion Atrovent* Combivent Respimat* ipratropium bromide ipratropium bromide 20mcg, 17mcg albuterol sulfate 100mcg Boehringer Ingelheim Boehringer Ingelheim Nebulized Albuterol Xopenex Inhalation Solution Xopenex Inhalation Solution Xopenex Inhalation Solution * Ipratropium bromide is not a recommended rescue inhaler albuterol sulfate levalbuterol HCl levalbuterol HCl levalbuterol HCl outside of use in the emergency room or urgent care but may, 2.5mg/3mL 0.31mg/3mL 0.63mg/3mL 1.25mg/3mL on occasion, be prescribed to supplement short-acting Beta 2 generic Sunovion Sunovion Sunovion agonists. Controllers Inhaled Corticosteroids (ICS): Metered-Dose Inhalers (MDI) Aerospan Alvesco Alvesco Asmanex Asmanex Flovent Flovent Flovent flunisolide ciclesonide ciclesonide mometasone furoate mometasone furoate fluticasone propionate fluticasone fluticasone propionate 80mcg 80mcg 160mcg 100mcg 200mcg 44mcg propionate 220mcg Meda Pharmaceuticals Sunovion Sunovion Merck Merck GlaxoSmithKline 110mcg GlaxoSmithKline GlaxoSmithKline Inhaled Corticosteroids (ICS): Dry Powder Inhalers(continued on back) QVAR QVAR beclomethasone beclomethasone dipropionate dipropionate 40mcg 80mcg ArmonAir RespiClick ArmonAir RespiClick ArmonAir RespiClick -

Beta Adrenergic Agents
Beta2 Adrenergic Agents –Long Acting Review 04/10/2008 Copyright © 2007 - 2008 by Provider Synergies, L.L.C. All rights reserved. Printed in the United States of America. All rights reserved. No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, recording, digital scanning, or via any information storage and retrieval system without the express written consent of Provider Synergies, L.L.C. All requests for permission should be mailed to: Attention: Copyright Administrator Intellectual Property Department Provider Synergies, L.L.C. 5181 Natorp Blvd., Suite 205 Mason, Ohio 45040 The materials contained herein represent the opinions of the collective authors and editors and should not be construed to be the official representation of any professional organization or group, any state Pharmacy and Therapeutics committee, any state Medicaid Agency, or any other clinical committee. This material is not intended to be relied upon as medical advice for specific medical cases and nothing contained herein should be relied upon by any patient, medical professional or layperson seeking information about a specific course of treatment for a specific medical condition. All readers of this material are responsible for independently obtaining medical advice and guidance from their own physician and/or other medical professional in regard to the best course of treatment for their specific medical condition. This publication, inclusive of all forms contained herein, is intended to be educational in nature and is intended to be used for informational purposes only. Comments and suggestions may be sent to [email protected]. -

The Use of Stems in the Selection of International Nonproprietary Names (INN) for Pharmaceutical Substances
WHO/PSM/QSM/2006.3 The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances 2006 Programme on International Nonproprietary Names (INN) Quality Assurance and Safety: Medicines Medicines Policy and Standards The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 © World Health Organization 2006 All rights reserved. Publications of the World Health Organization can be obtained from WHO Press, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel.: +41 22 791 3264; fax: +41 22 791 4857; e-mail: [email protected]). Requests for permission to reproduce or translate WHO publications – whether for sale or for noncommercial distribution – should be addressed to WHO Press, at the above address (fax: +41 22 791 4806; e-mail: [email protected]). The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate border lines for which there may not yet be full agreement. The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters. -

Long-Term Treatment of Chronic Obstructive Pulmonary Disease with Salmeterol and the Additive Effect of Ipratropium
Copyright #ERS Journals Ltd 2000 Eur Respir J 2000; 15: 878±885 European Respiratory Journal Printed in UK ± all rights reserved ISSN 0903-1936 Long-term treatment of chronic obstructive pulmonary disease with salmeterol and the additive effect of ipratropium J.A. van Noord*, D.R.A.J. de Munck**, Th.A. Bantje***, W.C.J. Hop+, M.L.M. Akveld++, A.M. Bommer++ Long-term treatment of chronic obstructive pulmonary disease with salmeterol and the Depts of Respiratory Diseases, *Atrium additive effect of ipratropium. J.A. van Noord, D.R.A.J. de Munck, Th.A. Bantje, W.C.J. medisch centrum, Heerlen, **St Joseph Hospital, Veldhoven and ***Ignatius Hos- Hop, M.L.M. Akveld, A.M. Bommer. #ERS Journals Ltd 2000. + ABSTRACT: The efficacy and safety of salmeterol alone was compared with the pital, Breda, Dept of Epidemiology and Biostatistics, Erasmus University, Rotter- combination of salmeterol plus ipratropium and with placebo during long-term dam and ++GlaxoWellcome, Zeist, the treatment in patients with stable chronic obstructive pulmonary disease. In addition, Netherlands. the single-dose effect in response to the first dose of treatment was studied over 12 h. The patients (n=144; age 647 yrs, forced expiratory volume in one second (FEV1) Correspondence: J.A. van Noord, Dept of 4411% pred) participated in a three-centre double-blind double-placebo parallel Respiratory Diseases, Atrium medisch group study and were randomized after a run-in period of 2 weeks to receive either centrum, Henri Dunantstraat 5, 6419 PC salmeterol 50 mg b.i.d., salmeterol 5 mg b.i.d. -

Skeletal and Cardiac Muscle Ergogenics and Side Effects Of
edicine s M & rt D o o p p i S n f g o S Douillard, J Sport Medic Doping Studie 2011, S1 l Journal of Sports Medicine & Doping t a u n d r i e u DOI: 10.4172/2161-0673.S1-001 s o J Studies ISSN: 2161-0673 Review Article Open Access Skeletal and Cardiac Muscle Ergogenics and Side Effects of Clenbuterol Treatment Aymeric Douillard* INRA, UMR866 Dynamique Musculaire et Métabolisme, Université Montpellier 1, F-34060 Montpellier, France Abstract Well known, well detected and still used by athletes, clenbuterol is one of the β2-agonists which has no authorization for therapeutic use, contrarily to salbutamol, salmeterol and formoterol in the 2012 World Anti-Doping Agency list. However, clenbuterol is still detected in athletes’ antidoping test samples. Its ability to induce muscle hypertrophy but also its strong lipolytic action and the absence of androgenic effects have made it a prized substances by athletes, specially females, without scruples whose performance requires significant muscle strength. Like the effects of clenbuterol on the heart, the effects of clenbuterol on skeletal muscle are dependent on the doses used and duration of the treatment. If there is a consensus concerning the clenbuterol action on the phenotypic conversion from slow to fast type fibers and on the hypertrophy, there is, to our knowledge, no consensus concerning the effects of clenbuterol on the slow type fibers and slow profile muscles. There is also no consensus concerning the clenbuterol effects on performance. We will shortly reviewing the known operating mode, side and benefits effects of short and long term β-agonists, and specially clenbuterol, treatment on mammals. -

Medicines for COPD
American Thoracic Society PATIENT EDUCATION | INFORMATION SERIES Medicines for COPD Chronic obstructive pulmonary disease (COPD) is a chronic disease of the lungs that damages both the airways and the lung tissue, making it difficult to breathe. A person with COPD may have obstructive bronchiolitis (bron-kee-oh-lite-is), emphysema, or a combination of both conditions. People with COPD often use several kinds of medicines to help control symptoms. While there is no cure, medicines can help improve your quality of life. This fact sheet explains different types of medicines used for COPD. For more information on COPD, see the ATS Patient Information Series at www.thoracic.org/patients. BRONCHODILATORS These side effects often last for only a few minutes after taking the Bronchodilators are medications that relax the muscles that wrap medicine and may totally go away after a few days of regular use. around your breathing tubes (airways), allowing the tubes to If the side effects do not go away, talk to your healthcare provider. become larger and easier to breathe through. Each bronchodilator You may need to try a lower dose, change to another type or brand is different, based on its: 1) chemical make-up, 2) how fast it works, of beta2-agonist, or stop the beta2-agonist. If you have difficulty and 3) how long it lasts. Your healthcare provider will decide with going to sleep after taking your beta2-agonist, take it an hour or you which of these medications or combinations work best for you. two before bedtime. Types of bronchodilators: Anticholinergics ■■ beta2-agonists Anticholinergic bronchodilators are also inhaled medicines. -

WO 2014/141069 Al 18 September 2014 (18.09.2014) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2014/141069 Al 18 September 2014 (18.09.2014) P O P C T (51) International Patent Classification: Novartis Pharmaceuticals Corporation, 150 Industrial A61K 9/00 (2006.01) A61K 31/00 (2006.01) Road, San Carlos, California 94070 (US). A61K 9/16 (2006.01) (74) Agent: MAZZA, Michael; Novartis Vaccines and Dia (21) International Application Number: gnostics, Inc., 4560 Horton Street, Emeryville, California PCT/IB2014/059632 94608 (US). (22) International Filing Date: (81) Designated States (unless otherwise indicated, for every 11 March 2014 ( 11.03.2014) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (25) Filing Language: English BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, (26) Publication Language: English DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, (30) Priority Data: KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, 61/784,865 14 March 2013 (14.03.2013) US MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, (71) Applicant (for all designated States except US) : NO¬ OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, VARTIS AG [CH/CH]; Lichtstrasse 35, CH-4056 Basel SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, (CH). -

2-Adrenergic Receptor Is Determined by Conformational Equilibrium in the Transmembrane Region
ARTICLE Received 21 Mar 2012 | Accepted 2 Aug 2012 | Published 4 Sep 2012 DOI: 10.1038/ncomms2046 Efficacy of theβ 2-adrenergic receptor is determined by conformational equilibrium in the transmembrane region Yutaka Kofuku1,2, Takumi Ueda1, Junya Okude1, Yutaro Shiraishi1, Keita Kondo1, Masahiro Maeda3, Hideki Tsujishita3 & Ichio Shimada1,4 Many drugs that target G-protein-coupled receptors (GPCRs) induce or inhibit their signal transduction with different strengths, which affect their therapeutic properties. However, the mechanism underlying the differences in the signalling levels is still not clear, although several structures of GPCRs complexed with ligands determined by X-ray crystallography are available. Here we utilized NMR to monitor the signals from the methionine residue at position 82 in neutral antagonist- and partial agonist-bound states of β2-adrenergic receptor (β2AR), which are correlated with the conformational changes of the transmembrane regions upon activation. We show that this residue exists in a conformational equilibrium between the inverse agonist- bound states and the full agonist-bound state, and the population of the latter reflects the signal transduction level in each ligand-bound state. These findings provide insights into the multi-level signalling of β2AR and other GPCRs, including the basal activity, and the mechanism of signal transduction mediated by GPCRs. 1 Graduate School of Pharmaceutical Sciences, The University of Tokyo, Hongo 7-3-1, Bunkyo-ku, Tokyo 113-0033, Japan. 2 Japan Biological Informatics Consortium (JBIC), Tokyo 135-0064, Japan. 3 Shionogi Co., Ltd., Discovery Research Laboratories, Osaka 561-0825, Japan. 4 Biomedicinal Information Research Center (BIRC), National Institute of Advanced Industrial Science and Technology (AIST), Aomi 2-41-6, Koto-ku, Tokyo 135-0064, Japan. -

PRAC Draft Agenda of Meeting 11-14 May 2020
11 May 2020 EMA/PRAC/257460/2020 Human Division Pharmacovigilance Risk Assessment Committee (PRAC) Draft agenda for the meeting on 11-14 May 2020 Chair: Sabine Straus – Vice-Chair: Martin Huber 11 May 2020, 10:30 – 19:30, via teleconference 12 May 2020, 08:30 – 19:30, via teleconference 13 May 2020, 08:30 – 19:30, via teleconference 14 May 2020, 08:30 – 16:00, via teleconference Organisational, regulatory and methodological matters (ORGAM) 28 May 2020, 09:00-12:00, via teleconference Disclaimers Some of the information contained in this agenda is considered commercially confidential or sensitive and therefore not disclosed. With regard to intended therapeutic indications or procedure scopes listed against products, it must be noted that these may not reflect the full wording proposed by applicants and may also change during the course of the review. Additional details on some of these procedures will be published in the PRAC meeting highlights once the procedures are finalised. Of note, this agenda is a working document primarily designed for PRAC members and the work the Committee undertakes. Note on access to documents Some documents mentioned in the agenda cannot be released at present following a request for access to documents within the framework of Regulation (EC) No 1049/2001 as they are subject to on-going procedures for which a final decision has not yet been adopted. They will become public when adopted or considered public according to the principles stated in the Agency policy on access to documents (EMA/127362/2006, Rev. 1). Official address Domenico Scarlattilaan 6 ● 1083 HS Amsterdam ● The Netherlands Address for visits and deliveries Refer to www.ema.europa.eu/how-to-find-us Send us a question Go to www.ema.europa.eu/contact Telephone +31 (0)88 781 6000 An agency of the European Union © European Medicines Agency, 2020. -
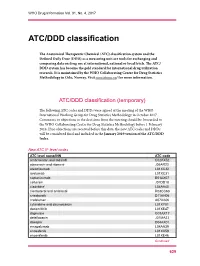
ATC/DDD Classification
WHO Drug Information Vol. 31, No. 4, 2017 ATC/DDD classification The Anatomical Therapeutic Chemical (ATC) classification system and the Defined Daily Dose (DDD) as a measuring unit are tools for exchanging and comparing data on drug use at international, national or local levels. The ATC/ DDD system has become the gold standard for international drug utilization research. It is maintained by the WHO Collaborating Centre for Drug Statistics Methodology in Oslo, Norway. Visit www.whocc.no/ for more information. ATC/DDD classification (temporary) The following ATC codes and DDDs were agreed at the meeting of the WHO International Working Group for Drug Statistics Methodology in October 2017. Comments or objections to the decisions from the meeting should be forwarded to the WHO Collaborating Centre for Drug Statistics Methodology before 1 February 2018. If no objections are received before this date, the new ATC codes and DDDs will be considered final and included in the January 2019 version of the ATC/DDD Index. New ATC 5th level codes ATC level name/INN ATC code ambrisentan and tadalafil C02KX52 atazanavir and ritonavir J05AR23 atezolizumab L01XC32 avelumab L01XC31 caplacizumab B01AX07 cefteram J01DD18 cladribine1 L04AA40 clenbuterol and ambroxol R03CC63 crisaborole D11AH06 crofelemer A07XA06 cytarabine and daunorubicin L01XY01 dacomitinib L01XE47 dapivirine G01AX17 delafloxacin J01MA23 doxepin D04AX01 emapalumab L04AA39 enasidenib L01XX59 encorafenib L01XE46 Continued 629 ATC/DDD classification WHO Drug Information Vol. 31, No. 4, 2017