Genetic Assessment of Boardman River Fish Populations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Research Funding (Total $2,552,481) $15,000 2019
CURRICULUM VITAE TENNESSEE AQUARIUM CONSERVATION INSTITUTE 175 BAYLOR SCHOOL RD CHATTANOOGA, TN 37405 RESEARCH FUNDING (TOTAL $2,552,481) $15,000 2019. Global Wildlife Conservation. Rediscovering the critically endangered Syr-Darya Shovelnose Sturgeon. $10,000 2019. Tennessee Wildlife Resources Agency. Propagation of the Common Logperch as a host for endangered mussel larvae. $8,420 2019. Tennessee Wildlife Resources Agency. Monitoring for the Laurel Dace. $4,417 2019. Tennessee Wildlife Resources Agency. Examining interactions between Laurel Dace (Chrosomus saylori) and sunfish $12,670 2019. Trout Unlimited. Southern Appalachian Brook Trout propagation for reintroduction to Shell Creek. $106,851 2019. Private Donation. Microplastic accumulation in fishes of the southeast. $1,471. 2019. AZFA-Clark Waldram Conservation Grant. Mayfly propagation for captive propagation programs. $20,000. 2019. Tennessee Valley Authority. Assessment of genetic diversity within Blotchside Logperch. $25,000. 2019. Riverview Foundation. Launching Hidden Rivers in the Southeast. $11,170. 2018. Trout Unlimited. Propagation of Southern Appalachian Brook Trout for Supplemental Reintroduction. $1,471. 2018. AZFA Clark Waldram Conservation Grant. Climate Change Impacts on Headwater Stream Vertebrates in Southeastern United States $1,000. 2018. Hamilton County Health Department. Step 1 Teaching Garden Grants for Sequoyah School Garden. $41,000. 2018. Riverview Foundation. River Teachers: Workshops for Educators. $1,000. 2018. Tennessee Valley Authority. Youth Freshwater Summit $20,000. 2017. Tennessee Valley Authority. Lake Sturgeon Propagation. $7,500 2017. Trout Unlimited. Brook Trout Propagation. $24,783. 2017. Tennessee Wildlife Resource Agency. Assessment of Percina macrocephala and Etheostoma cinereum populations within the Duck River Basin. $35,000. 2017. U.S. Fish and Wildlife Service. Status surveys for conservation status of Ashy (Etheostoma cinereum) and Redlips (Etheostoma maydeni) Darters. -

Forecasting the Impacts of Silver and Bighead Carp on the Lake Erie Food Web
Transactions of the American Fisheries Society ISSN: 0002-8487 (Print) 1548-8659 (Online) Journal homepage: http://www.tandfonline.com/loi/utaf20 Forecasting the Impacts of Silver and Bighead Carp on the Lake Erie Food Web Hongyan Zhang, Edward S. Rutherford, Doran M. Mason, Jason T. Breck, Marion E. Wittmann, Roger M. Cooke, David M. Lodge, John D. Rothlisberger, Xinhua Zhu & Timothy B. Johnson To cite this article: Hongyan Zhang, Edward S. Rutherford, Doran M. Mason, Jason T. Breck, Marion E. Wittmann, Roger M. Cooke, David M. Lodge, John D. Rothlisberger, Xinhua Zhu & Timothy B. Johnson (2016) Forecasting the Impacts of Silver and Bighead Carp on the Lake Erie Food Web, Transactions of the American Fisheries Society, 145:1, 136-162, DOI: 10.1080/00028487.2015.1069211 To link to this article: http://dx.doi.org/10.1080/00028487.2015.1069211 © 2016 The Author(s). Published with View supplementary material license by American Fisheries Society© Hongyan Zhang, Edward S. Rutherford, Doran M. Mason, Jason T. Breck, Marion E. Wittmann, Roger M. Cooke, David M. Lodge, Published online: 30 Dec 2015. Submit your article to this journal John D. Rothlisberger, Xinhua Zhu, Timothy B. Johnson Article views: 1095 View related articles View Crossmark data Full Terms & Conditions of access and use can be found at http://www.tandfonline.com/action/journalInformation?journalCode=utaf20 Download by: [University of Strathclyde] Date: 02 March 2016, At: 02:30 Transactions of the American Fisheries Society 145:136–162, 2016 Published with license by American Fisheries Society 2016 ISSN: 0002-8487 print / 1548-8659 online DOI: 10.1080/00028487.2015.1069211 ARTICLE Forecasting the Impacts of Silver and Bighead Carp on the Lake Erie Food Web Hongyan Zhang* Cooperative Institute for Limnology and Ecosystems Research, School of Natural Resources and Environment, University of Michigan, 4840 South State Road, Ann Arbor, Michigan 48108, USA Edward S. -

On the Niagara Peninsula, Canada
Genetic evidence for canal-mediated dispersal of Mapleleaf, Quadrula quadrula (Bivalvia:Unionidae) on the Niagara Peninsula, Canada Jordan R. Hoffman1,3, Todd J. Morris2,4, and David T. Zanatta1,5 1Institute for Great Lakes Research, Biology Department, Central Michigan University, Mount Pleasant, Michigan 48859 USA 2Great Lakes Laboratory for Fisheries and Aquatic Sciences, Fisheries and Oceans Canada, Burlington, Ontario, Canada L7S 1A1 Abstract: Alterations to watercourses affect connectivity in aquatic systems and can influence dispersal of aquatic biota. Dams fragment populations and act as isolating barriers, but canals create connections between waterbodies that can be used as corridors for dispersal by opportunistic invaders. The Niagara Peninsula of Ontario, Canada, has a 200-y history of canal operation, resulting in major modification of the watercourses in the region. This mod- ification allowed numerous invasive species to enter the upper Great Lakes (e.g., sea lamprey) and probably has facilitated dispersal in native species. The purpose of our study was to explore the effects of canal and dam con- struction on the genetic structure of Mapleleaf (Quadrula quadrula), a widespread and relatively common species in the central Great Lakes that has been found only recently in several western Lake Ontario harbors. Establish- ment of Q. quadrula in Lake Ontario may have been a recent event, facilitated by the Niagara Peninsula’s history of canal operation. We used analyses of microsatellite DNA genotypes to examine the effect of canals on the genetic structure of mussel populations. Structure analysis revealed a pattern of gene flow between lakes that cannot be explained by watercourse connections prior to the creation of the Welland Canal. -

Guiding Species Recovery Through Assessment of Spatial And
Guiding Species Recovery through Assessment of Spatial and Temporal Population Genetic Structure of Two Critically Endangered Freshwater Mussel Species (Bivalvia: Unionidae) Jess Walter Jones ( [email protected] ) United States Fish and Wildlife Service Timothy W. Lane Virginia Department of Game and Inland Fisheries N J Eric M. Hallerman Virginia Tech: Virginia Polytechnic Institute and State University Research Article Keywords: Freshwater mussels, Epioblasma brevidens, E. capsaeformis, endangered species, spatial and temporal genetic variation, effective population size, species recovery planning, conservation genetics Posted Date: March 16th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-282423/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/28 Abstract The Cumberlandian Combshell (Epioblasma brevidens) and Oyster Mussel (E. capsaeformis) are critically endangered freshwater mussel species native to the Tennessee and Cumberland River drainages, major tributaries of the Ohio River in the eastern United States. The Clinch River in northeastern Tennessee (TN) and southwestern Virginia (VA) harbors the only remaining stronghold population for either species, containing tens of thousands of individuals per species; however, a few smaller populations are still extant in other rivers. We collected and analyzed genetic data to assist with population restoration and recovery planning for both species. We used an 888 base-pair sequence of the mitochondrial NADH dehydrogenase 1 (ND1) gene and ten nuclear DNA microsatellite loci to assess patterns of genetic differentiation and diversity in populations at small and large spatial scales, and at a 9-year (2004 to 2013) temporal scale, which showed how quickly these populations can diverge from each other in a short time period. -

GCP LCC Regional Hypotheses of Ecological Responses to Flow
Gulf Coast Prairie Landscape Conservation Cooperative Regional Hypotheses of Ecological Responses to Flow Alteration Photo credit: Brandon Brown A report by the GCP LCC Flow-Ecology Hypotheses Committee Edited by: Mary Davis, Coordinator, Southern Aquatic Resources Partnership 3563 Hamstead Ct, Durham, North Carolina 27707, email: [email protected] and Shannon K. Brewer, U.S. Geological Survey Oklahoma Cooperative Fish and Wildlife Research Unit, 007 Agriculture Hall, Stillwater, Oklahoma 74078 email: [email protected] Wildlife Management Institute Grant Number GCP LCC 2012-003 May 2014 ACKNOWLEDGMENTS We thank the GCP LCC Flow-Ecology Hypotheses Committee members for their time and thoughtful input into the development and testing of the regional flow-ecology hypotheses. Shannon Brewer, Jacquelyn Duke, Kimberly Elkin, Nicole Farless, Timothy Grabowski, Kevin Mayes, Robert Mollenhauer, Trevor Starks, Kevin Stubbs, Andrew Taylor, and Caryn Vaughn authored the flow-ecology hypotheses presented in this report. Daniel Fenner, Thom Hardy, David Martinez, Robby Maxwell, Bryan Piazza, and Ryan Smith provided helpful reviews and improved the quality of the report. Funding for this work was provided by the Gulf Coastal Prairie Landscape Conservation Cooperative of the U.S. Fish and Wildlife Service and administered by the Wildlife Management Institute (Grant Number GCP LCC 2012-003). Any use of trade, firm, or product names is for descriptive purposes and does not imply endorsement by the U.S. Government. Suggested Citation: Davis, M. M. and S. Brewer (eds.). 2014. Gulf Coast Prairie Landscape Conservation Cooperative Regional Hypotheses of Ecological Responses to Flow Alteration. A report by the GCP LCC Flow-Ecology Hypotheses Committee to the Southeast Aquatic Resources Partnership (SARP) for the GCP LCC Instream Flow Project. -

Leptodea Leptodon
Natural Resource Ecology and Management Natural Resource Ecology and Management Publications 2017 A comparison of genetic diversity and population structure of the endangered scaleshell mussel (Leptodea leptodon), the fragile papershell (Leptodea fragilis) and their host-fish the freshwater drum (Aplodinotus grunniens) Jer Pin Chong The University of Illinois at Chicago Kevin J. Roe Iowa State University, [email protected] Follow this and additional works at: https://lib.dr.iastate.edu/nrem_pubs Part of the Ecology and Evolutionary Biology Commons, Genetics Commons, Natural Resources and Conservation Commons, and the Natural Resources Management and Policy Commons The ompc lete bibliographic information for this item can be found at https://lib.dr.iastate.edu/ nrem_pubs/264. For information on how to cite this item, please visit http://lib.dr.iastate.edu/ howtocite.html. This Article is brought to you for free and open access by the Natural Resource Ecology and Management at Iowa State University Digital Repository. It has been accepted for inclusion in Natural Resource Ecology and Management Publications by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. A comparison of genetic diversity and population structure of the endangered scaleshell mussel (Leptodea leptodon), the fragile papershell (Leptodea fragilis) and their host-fish the freshwater drum (Aplodinotus grunniens) Abstract The al rvae of freshwater mussels in the order Unionoida are obligate parasites on fishes. Because adult mussels are infaunal and largely sessile, it is generally assumed that the majority of gene flow among mussel populations relies on the dispersal of larvae by their hosts. -

The Influence of Flow Alteration on Instream Habitat and Fish Assemblages
THE INFLUENCE OF FLOW ALTERATION ON INSTREAM HABITAT AND FISH ASSEMBLAGES By NICOLE FARLESS Bachelor of Science in Agriculture Sciences and Natural Resources Oklahoma State University Stillwater, Oklahoma 2012 Submitted to the Faculty of the Graduate College of the Oklahoma State University in partial fulfillment of the requirements for the Degree of MASTER OF SCIENCE December, 2015 THE INFLUENCE OF FLOW ALTERATION ON INSTREAM HABITAT AND FISH ASSEMBLAGES Thesis Approved: Shannon Brewer Thesis Adviser Todd Halihan Jim Long ii ACKNOWLEDGEMENTS There are many people that I need to acknowledge for their help and support throughout my Master’s research. I would like to start off thanking my advisor, Dr. Shannon Brewer. Shannon not only continually helped me broaden my knowledge of science and research but also invested her time to improve my practical knowledge as well. I would also like to thank my funding source, The Nature Conservancy and the Oklahoma Cooperative Fish and Wildlife Research Unit. I would also like to thank Brian Brewer for all of his help building and developing the thermal tolerance lab. Without him I could not have completed my temperature tolerance study. I am indebted to many graduate students that have assisted with statistical, theoretical, and field work. Especially, Jonathan Harris and Robert Mollenhauer, who have volunteered countless hours editing my writing and assisting with data analysis. I would also like to thank Dr. Jim Shaw, my undergraduate advisor, for recognizing my potential and encouraging me to attend graduate school. Without him I would have never considered furthering my education. I would not have been able to complete this project without the help of the many technicians that worked hard for me in the lab and in the field: Desiree Williams, Frances Marshall, Joshua Mouser, Bailey Johnson, Emily Gardner, Spencer Wood, Cooper Sherrill, and Jake Holliday. -

Do the Invasive Fish Species Round Goby (Neogobius Melanostomus) and the Native Fish Species Viviparous Eelpout (Zoarces Viviparus) Compete for Shelter?
Thea Bjørnsdatter Gullichsen Do the invasive fish species round goby (Neogobius melanostomus) and the native fish species viviparous eelpout (Zoarces viviparus) compete for shelter? Master’s thesis Master’s Master’s thesis in Biology Supervisor: Gunilla Rosenqvist, Irja Ida Ratikainen, Isa Wallin May 2019 NTNU Department of Biology Faculty of Natural Sciences of Natural Faculty Norwegian University of Science and Technology of Science University Norwegian Thea Bjørnsdatter Gullichsen Do the invasive fish species round goby (Neogobius melanostomus) and the native fish species viviparous eelpout (Zoarces viviparus) compete for shelter? Master’s thesis in Biology Supervisor: Gunilla Rosenqvist, Irja Ida Ratikainen, Isa Wallin May 2019 Norwegian University of Science and Technology Faculty of Natural Sciences Department of Biology Abstract Human-mediated introduction of species has increased drastically the last decades, with the increased connection across borders. The settlement of invasive species in a new environment can result in drastic changes in the ecosystem, and even result in local extinction of native species. One species that is currently regarded one of the most invasive species in the Baltic Sea is the round goby (Neogobius melanostomus). Round goby and the native species viviparous eelpout (Zoarces viviparus) are both benthic dwellers that inhabits the coast of Gotland in Sweden. With a shared habitat and the round goby being a highly competitive species, it can be expected that the viviparous eelpout is affected negatively by the round goby in some way. I performed a laboratory study to determine if round goby and viviparous eelpout compete for shelter when sharing a fish tank. I predicted that the round goby would guard the shelter by demonstrating aggressive behaviour when paired with the viviparous eelpout. -

Biological Assessment for Mountain Valley Pipeline, Llc Mountain Valley Pipeline Project Ferc Docket No
20170707-4008 FERC PDF (Unofficial) 07/07/2017 BIOLOGICAL ASSESSMENT FOR MOUNTAIN VALLEY PIPELINE, LLC MOUNTAIN VALLEY PIPELINE PROJECT FERC DOCKET NO. CP16-10-000 20170707-4008 FERC PDF (Unofficial) 07/07/2017 This page intentionally left blank. 20170707-4008 FERC PDF (Unofficial) 07/07/2017 TABLE OF CONTENTS 1.0 Executive Summary ....................................................................................................... 1-1 2.0 Introduction .................................................................................................................... 2-1 2.1 ESA Species of Concern .................................................................................... 2-2 2.2 Consultation History .......................................................................................... 2-4 3.0 Description of the Proposed Action .............................................................................. 3-1 3.1 Construction ....................................................................................................... 3-1 3.1.1 Pipeline .................................................................................................... 3-1 3.1.2 Aboveground Facilities ............................................................................ 3-3 3.1.3 Yards ........................................................................................................ 3-8 3.1.4 Access Roads ........................................................................................... 3-8 3.1.5 Additional Temporary Workspaces -

Final Fisheries Existing Data
BOARDMAN RIVER FEASIBILITY STUDY A Report on the Boardman River Fisheries Existing Data January 2009 Submitted by: 2200 Commonwealth Blvd, Suite 300 Ann Arbor, MI 48105 Ph: 734-769-3004 Fax: 734-769-3164 ACKNOWLEDGMENTS We would like to acknowledge the U.S. Department of Interior, Fish and Wildlife Service, for funding this important work through a Tribal Wildlife Grant provided to the Grand Traverse Band of Ottawa and Chippewa Indians. Finally, the project team would like to thank the following organizations for their assistance and for providing critical data that was used in the preparation of this report: Grand Traverse Band of Ottawa and Chippewa Indians (GTB) Michigan Department of Natural Resources – Fish Division (MDNR) Michigan Department of Environmental Quality – Water Bureau (MDEQ) Michigan State University, Department of Fisheries & Wildlife (MSUFW) United States Fish and Wildlife Service (USFWS), Watershed Center Grand Traverse Bay (WCGTB) ECT Project Team: Dr. Bryan Burroughs, Michigan State University (Primary author of this document) Dr. Sanjiv Sinha, Ph.D. - Environmental Consulting & Technology, Inc. (Key Technical Resource) Dr. Donald Tilton, Ph.D. - Environmental Consulting & Technology, Inc. (Project Manager) Mr. Scott Parker, Environmental Consulting & Technology, Inc. (Project Director) Roy Schrameck, Environmental Consulting & Technology, Inc. (Environmental Team Lead) Boardman River Feasibility Study – A Report on the Boardman River Fisheries Existing Data TABLE OF CONTENTS Section Page Purpose ....................................................................................................................................................... -

Boardman River Mainstem Grand Traverse County, Michigan
Section 506 Great Lakes Fishery and Ecosystem Restoration Preliminary Restoration Plan for the Boardman River Mainstem Grand Traverse County, Michigan November 2005 U.S. Army Corps of Engineers Boardman River PRP DRAFT Preliminary Restoration Plan Boardman River Section 506 Great Lakes Fishery and Ecosystem Restoration 1. PROJECT: Boardman River: Great Lakes Fishery and Ecosystem Restoration Congressional District: MI-4, Camp Senators Stabenow and Levin Authority: Section 506 (Great Lakes Fishery and Ecosystem Restoration) of the Water Resources Development Act of 2000 (PL 106-541), which states in paragraph c(2): “The Secretary shall plan, design, and construct projects to support the restoration of the fishery, ecosystem, and beneficial uses of the Great Lakes”. 2. LOCATION: State: Michigan County: Grand Traverse The Boardman River is located in the northwestern portion of Michigan’s Lower Peninsula. The river originates in central Kalkaska County, flows southwest into Grand Traverse County where it turns north and flows into West Grand Traverse Bay, Lake Michigan in Traverse City, Michigan (Figures 1 and 2). The Boardman River watershed drains a surface area of approximately 291 square miles and includes 179 lineal stream miles and 12 natural lakes. The project area is a 20-mile plus section of the Boardman River’s main stem, located in Grand Traverse County, which empties into the bay and contains four dams (Figure 3): Union Street Dam, located at river mile 1.5, Sabin Dam at 5.3, Boardman Dam at 6.1, and Brown Bridge Dam at 18.5. The project area spans from upstream of Brown Bridge Pond’s inlet to the mouth of the river. -
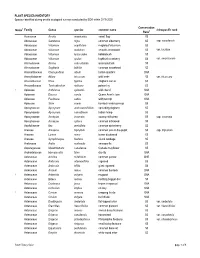
Appendix 11-D Plant and Wildlife Species Lists
PLANT SPECIES INVENTORY Species identified during on-site ecological surveys conducted by EDR within 2019-2020 Conservation Notes1 Family Genus species common name infraspecific rank Rank2 Acoraceae Acorus americanus sweet flag S5 Adoxaceae Sambucus nigra common elderberry S5 ssp. canadensis Adoxaceae Viburnum acerifolium mapleleaf viburnum S5 Adoxaceae Viburnum dentatum smooth arrowwood S5 var. lucidum Adoxaceae Viburnum lantanoides hobblebush S5 Adoxaceae Viburnum opulus highbush cranberry S4 var. americanum Alismataceae Alisma subcordatum water-plantain S5 Alismataceae Sagittaria latifolia common arrowhead S5 Amaranthaceae Chenopodium album lambs-quarters SNA Amaryllidaceae Allium tricoccum wild leeks S5 var. tricoccum Anacardiaceae Rhus typhina staghorn sumac S5 Anacardiaceae Toxicodendron radicans poison ivy S5 1 Apiaceae Anthriscus sylvestris wild chervil SNA Apiaceae Daucus carota Queen Anne's lace SNA Apiaceae Pastinaca sativa wild parsnip SNA Apiaceae Sium suave hemlock water-parnsip S5 Apocynaceae Apocynum androsaemifolium spreading dogbane S5 Apocynaceae Apocynum cannabinum Indian hemp S5 Apocynaceae Asclepias incarnata swamp milkweed S5 ssp. incarnata Apocynaceae Asclepias syriaca common milkweed S5 Aquifoliaceae Ilex verticillata common winterberry S5 Araceae Arisaema triphyllum common jack-in-the-pulpit S5 ssp. triphyllum Araceae Lemna minor lesser duckweed S5 Araceae Symplocarpus foetidus skunk cabbage S5 Araliaceae Aralia nudicaulis sarsaparilla S5 Asparagaceae Maianthemum canadense Canada mayflower S5 Asphodelaceae Hemerocallis