Deep Learning for Single-Molecule Science
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Fundamental Sensitivity Limit Imposed by Dark 1/F Noise in the Low Optical Signal Detection Regime
Fundamental sensitivity limit imposed by dark 1/f noise in the low optical signal detection regime Emily J. McDowell1, Jian Ren2, and Changhuei Yang1,2 1Department of Bioengineering, 2Department of Electrical Engineering, Mail Code 136-93,California Institute of Technology, Pasadena, CA, 91125 [email protected] Abstract: The impact of dark 1/f noise on fundamental signal sensitivity in direct low optical signal detection is an understudied issue. In this theoretical manuscript, we study the limitations of an idealized detector with a combination of white noise and 1/f noise, operating in detector dark noise limited mode. In contrast to white noise limited detection schemes, for which there is no fundamental minimum signal sensitivity limit, we find that the 1/f noise characteristics, including the noise exponent factor and the relative amplitudes of white and 1/f noise, set a fundamental limit on the minimum signal that such a detector can detect. © 2008 Optical Society of America OCIS codes: (230.5160) Photodetectors; (040.3780) Low light level References and links 1. S. O. Flyckt and C. Marmonier, Photomultiplier tubes: Principles and applications (Photonis, 2002). 2. M. C. Teich, K. Matsuo, and B. E. A. Saleh, "Excess noise factors for conventional and superlattice avalanche photodiodes and photomultiplier tubes," IEEE J. Quantum Electron. 22, 1184-1193 (1986). 3. E. J. McDowell, X. Cui, Y. Yaqoob, and C. Yang, "A generalized noise variance analysis model and its application to the characterization of 1/f noise," Opt. Express 15, (2007). 4. P. Dutta and P. M. Horn, "Low-frequency fluctuations in solids - 1/f noise," Rev. -

Boundary-Preserved Deep Denoising of the Stochastic Resonance Enhanced Multiphoton Images
Boundary-Preserved Deep Denoising of the Stochastic Resonance Enhanced Multiphoton Images SHENG-YONG NIU1,2, LUN-ZHANG GUO3, YUE LI4, TZUNG-DAU WANG5, YU TSAO1,*, TZU-MING LIU4,* 1. Research Center for Information Technology Innovation (CITI), Academia Sinica, Taipei, Taiwan. 2. Department of Computer Science and Engineering, University of California San Diego, CA, USA. 3. Institute of Biomedical Engineering, National Taiwan University, Taipei 10617, Taiwan. 4. Institute of Translational Medicine, Faculty of Health Sciences, University of Macau, Macao SAR, China. 5. Cardiovascular Center and Division of Cardiology, Department of Internal Medicine, National Taiwan University Hospital and College of Medicine, Taipei 10002, Taiwan. * [email protected] and [email protected] Abstract: As the rapid growth of high-speed and deep-tissue imaging in biomedical research, it is urgent to find a robust and effective denoising method to retain morphological features for further texture analysis and segmentation. Conventional denoising filters and models can easily suppress perturbative noises in high contrast images. However, for low photon budget multi- photon images, high detector gain will not only boost signals, but also bring huge background noises. In such stochastic resonance regime of imaging, sub-threshold signals may be detectable with the help of noises. Therefore, a denoising filter that can smartly remove noises without sacrificing the important cellular features such as cell boundaries is highly desired. In this paper, we propose a convolutional neural network based denoising autoencoder method, Fully Convolutional Deep Denoising Autoencoder (DDAE), to improve the quality of Three-Photon Fluorescence (3PF) and Third Harmonic Generation (THG) microscopy images. The average of the acquired 200 images of a given location served as the low-noise answer for DDAE training. -
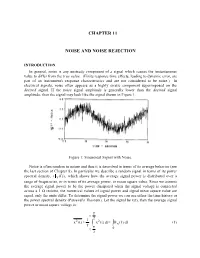
Chapter 11 Noise and Noise Rejection
CHAPTER 11 NOISE AND NOISE REJECTION INTRODUCTION In general, noise is any unsteady component of a signal which causes the instantaneous value to differ from the true value. (Finite response time effects, leading to dynamic error, are part of an instrument's response characteristics and are not considered to be noise.) In electrical signals, noise often appears as a highly erratic component superimposed on the desired signal. If the noise signal amplitude is generally lower than the desired signal amplitude, then the signal may look like the signal shown in Figure 1. Figure 1: Sinusoidal Signal with Noise. Noise is often random in nature and thus it is described in terms of its average behavior (see the last section of Chapter 8). In particular we describe a random signal in terms of its power spectral density, (x (f )) , which shows how the average signal power is distributed over a range of frequencies, or in terms of its average power, or mean square value. Since we assume the average signal power to be the power dissipated when the signal voltage is connected across a 1 Ω resistor, the numerical values of signal power and signal mean square value are equal, only the units differ. To determine the signal power we can use either the time history or the power spectral density (Parseval's Theorem). Let the signal be x(t), then the average signal power or mean square voltage is: T t 2 221 x(t) x(t)dtx (f)df (1) T T t 0 2 11-2 Note: the bar notation, , denotes a time average taken over many oscillations of the signal. -

Grant County Planning and Zoning on WES
Bald and Golden Eagles, Grant and Codington Counties, 2017 & 2018 Grant County Planning and Zoning on WES April 17th, 2018 1. Questions from the public 2. General information and where to find more information, includes setback information from Vesta manuals, health, decommissioning and return on Tax Production Credit 3. Health information and where to find more, includes Cooper interview and personal testimony 4. Property value loses, includes report from PUC debunking the Next Era claim 5. Decommissioning issues 6. Language used in Wind Contracts This book contains websites and where to find more information. If you have questions, need additional reports you may contact Vince Meyer 605-949-1916 Facts, Findings and Conclusions We would like FINDINGS to the following issues to be addressed as answers in local newspapers for the public to review and discuss. 1. Has Next Era, APEX, or any wind energy developer, provided scientific methods to prove flicker, sound, infrasound or other nuisance, will trespass on to non-participants' land, for a certain allotment of time, please present the methods and studies to support the claim. 2. Have Planning and Zoning members and Commissioners read a Turbine manual for all the models being proposed to be used for safety setbacks? If so, where is a manual for the public to evaluate? 3. Has a MSDS for the county? 4. Wind farms are privately owned. They do not supply electricity to the public. Their electricity is for sale. Hence they are not a public utility. How will the county or township handle trespassing on private land be it rights-of-ways or other types of property? 5. -
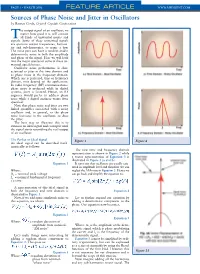
Sources of Phase Noise and Jitter in Oscillators by Ramon Cerda, Crystek Crystals Corporation
PAGE • MARCH 2006 FEATURE ARTICLE WWW.MPDIGEST.COM Sources of Phase Noise and Jitter in Oscillators by Ramon Cerda, Crystek Crystals Corporation he output signal of an oscillator, no matter how good it is, will contain Tall kinds of unwanted noises and signals. Some of these unwanted signals are spurious output frequencies, harmon- ics and sub-harmonics, to name a few. The noise part can have a random and/or deterministic noise in both the amplitude and phase of the signal. Here we will look into the major sources of some of these un- wanted signals/noises. Oscillator noise performance is char- acterized as jitter in the time domain and as phase noise in the frequency domain. Which one is preferred, time or frequency domain, may depend on the application. In radio frequency (RF) communications, phase noise is preferred while in digital systems, jitter is favored. Hence, an RF engineer would prefer to address phase noise while a digital engineer wants jitter specified. Note that phase noise and jitter are two linked quantities associated with a noisy oscillator and, in general, as the phase noise increases in the oscillator, so does the jitter. The best way to illustrate this is to examine an ideal signal and corrupt it until the signal starts resembling the real output of an oscillator. The Perfect or Ideal Signal Figure 1 Figure 2 An ideal signal can be described math- ematically as follows: The new time and frequency domain representation is shown in Figure 2 while a vector representation of Equation 3 is illustrated in Figure 3 (a and b.) Equation 1 It turns out that oscillators are usually satu- rated in amplitude level and therefore we can Where: neglect the AM noise in Equation 3. -

MT-048: Op Amp Noise Relationships
MT-048 TUTORIAL Op Amp Noise Relationships: 1/f Noise, RMS Noise, and Equivalent Noise Bandwidth "1/f" NOISE The general characteristic of op amp current or voltage noise is shown in Figure 1 below. NOISE 3dB/Octave 1 nV / √Hz e , i = k F n n C f or μV / √Hz 1 CORNER f en, in WHITE NOISE k F LOG f C Figure 1: Frequency Characteristic of Op Amp Noise At high frequencies the noise is white (i.e., its spectral density does not vary with frequency). This is true over most of an op amp's frequency range, but at low frequencies the noise spectral density rises at 3 dB/octave, as shown in Figure 1 above. The power spectral density in this region is inversely proportional to frequency, and therefore the voltage noise spectral density is inversely proportional to the square root of the frequency. For this reason, this noise is commonly referred to as 1/f noise. Note however, that some textbooks still use the older term flicker noise. The frequency at which this noise starts to rise is known as the 1/f corner frequency (FC) and is a figure of merit—the lower it is, the better. The 1/f corner frequencies are not necessarily the same for the voltage noise and the current noise of a particular amplifier, and a current feedback op amp may have three 1/f corners: for its voltage noise, its inverting input current noise, and its non-inverting input current noise. The general equation which describes the voltage or current noise spectral density in the 1/f region is 1 = Fk,i,e , Eq. -

Flicker Noise of Phase in Rf Amplifiers and Frequency Multipliers
FLICKER NOISE OF PHASE IN RF AMPLIFIERS AND FREQUENCYMULTIPLIERS: CHARACTERIZATION, CAUSE, AND CURE Donald Halford, A. E.Wainwright, and James A. Barnes Time and Frequency Division National Bureau of Standards, Boulder, Colorado 80302 Summary The high phase stability of atomic frequency standards has called for the development of associated electronic equipment of equivalent or superior stability. We have surveyed the performance of existing high quality frequency multipliers and RF amplifiers which operate in the range of 5 MHz to microwave frequencies. We were most interested in the phase noise in the range of Fourierfrequencies, f, of about Hz to lot3 Hz, sincemost electronic servo systems in existing atomic frequency standards use modula- tion frequencies which fall within this range. This range also includes the passive linewidths of existing atomic frequency standards. We found that all state-of-the-art, solid state amplifiers and frequency multipliers in our survey had random phase fluctuations (phase noise) of significant intensity and witha spectral density proportional to l/f (flicker noise of phase). Surprisingly, the flicker noise of phase was found to be approximately the same in most of the apparatus included in the survey. The typical one-sided spectral density of the phase noise was about (10- radian2)/f when referred to the input frequency, and the best performance was only 6 dB better. This performance was independent of the input fre- quency over at least the surveyed range of 5 MHz to 100 MHz, and did not depend upon the multiplication factor. This noise level exceeds by about 20 dB the stability which is needed for full compatibility with existing hydro- gen atom masers and with proposed high flux cesium beam designs. -

Flicker Noise Formulations in Compact Models Geoffrey J
This article has been accepted for publication in a future issue of this journal, but has not been fully edited. Content may change prior to final publication. Citation information: DOI 10.1109/TCAD.2020.2966444, IEEE Transactions on Computer-Aided Design of Integrated Circuits and Systems 1 Flicker Noise Formulations in Compact Models Geoffrey J. Coram, Senior Member, IEEE, Colin C. McAndrew, Fellow, IEEE, Kiran K. Gullapalli, and Kenneth S. Kundert, Fellow, IEEE Abstract—This paper shows how to properly define modulated flicker noise in Verilog-A compact models, and how a simulator should handle flicker noise for periodic and transient analyses. stationary noise By considering flicker noise in a simple linear resistor driven S(f) cyclostationary noise by a sinusoidal source, we demonstrate that the absolute value formulation used in most existing Verilog-A flicker noise models is incorrect when the bias applied to the resistor changes sign. m(t) Our new method for definition overcomes this problem, in the resistor as well as more sophisticated devices. The generalization of our approach should be adopted for flicker noise, replacing the formulation in existing Verilog-A device models, and it should be used in all new models. Since Verilog-A is the de facto Fig. 1. The creation of cyclostationary noise by a periodically-varying bias standard language for compact modeling, it is critical that model m(t) in a modulated stationary noise model. developers use the correct formulation. Index Terms—Flicker noise, compact models, Verilog-A, mod- ulated stationary noise model. these approaches allow accounting for frequency translation of noise, an effect that is not obtained from standard ac noise analysis [7]. -

Study and Development of Low-Noise MEMS Acoustic Sensors
OMRON TECHNICS Vol.50.012EN 2019.3 Study and development of low-noise MEMS acoustic sensors Important considerations for air damping and process stability INOUE Tadashi, UDHIDA Yuki, ISHIMOTO Koichi and HORIMOTO Yasuhiro Acoustic sensing technology like speech recognition or noise cancellation has attracted a lot of attention recently. These new applications stimulate the demand for smaller and lower noise MEMS (micro electro-mechanical system) acoustic sensors. Micro-scale sensors are susceptible to self-noise caused by air damping. Therefore, understanding and controlling air damping is crucial for designing low noise sensors. In this paper, we report a novel design of MEMS acoustic sensors that achieves SNR (signal-to-noise ratio) over 68 dB. We simulated dominant noise sources based on equivalent circuit analysis and introduced original structures that significantly reduce squeeze air film damping. This new design has been successfully commercialized due to matured process stability techniques of thin-film. The acoustic sensors we developed can be widely used in applications that require small-scale and precise acoustic sensing. 1. Introduction subject. As a result, we succeeded in achieving significantly lower A trend toward mounting various sensors on various instruments noise than at the time of the beginning of mass production by or living organisms to collect data and use the obtained data for identifying major noise sources and redesigning the corresponding livelihoods and industries has been growing day by day. Under structures. In this paper, we report on the simulation aimed at such circumstances, expectations are high for small and high- the reduction of noise, the result of the verification of the noise precision sensor devices. -

Journal La Multiapp
JOURNAL LA MULTIAPP VOL. 01, ISSUE 05(025-027), 2020 DOI: 10.37899/journallamultiapp.v1i5.275 Noise in Telecommunication: Different Types and Methods of dealing with Noise Akramjon Mirzaev1, Sanjar Zoteev1 1Telecommunication Engineering Department, Tashkent University of Information Technologies, Uzbekistan *Corresponding Author: Akramjon Mirzaev Article Info Abstract Article history: This article discusses noise in telecommunications: different types and Received 5 December 2020 methods of dealing with noise. Noise is arguably a very hated problem Received in revised form 25 because it can interfere with the quality of signal reception and also December 2020 the reproduction of the signal that will be transmitted. Not only that, Accepted 31 December 2020 but noise can also limit the range of the system to a certain emission power and can affect the sensitivity and sensitivity of the reception Keywords: signal. Even in some cases, noise can also result in a reduction in the Telecommunication bandwidth of a system. Of course, we've all felt how annoying the noise Noise effect is. For example, when listening to the radio, a hissing sound Performance appears on the loudspeaker due to noise. To overcome noise, it is divided into passive noise control and active noise control. Passive noise control is an effort to overcome noise using components that do not require power. Generally passive noise control uses soundproof materials that act as insulation against noise. The method most commonly used to overcome noise is through increasing the gain. The noise is generally in a specific sound area. Hiss is on high frequencies, while noise and hum are on low frequencies. -

Study of Flicker Noise for Zero-IF Receiver
Progress In Electromagnetics Research Symposium 2005, Hangzhou, China, August 22-26 591 Study of Flicker Noise for Zero-IF Receiver Jun Gao, Jinsheng Tang, and Kemin Sheng Southwest Jiaotong University, China Abstract Zero-IF receiver has some advantages, such as small factor, low cost and easily integrated on a chip. They make it competitive of RF receivers. However, DC Offset and flicker noise, etc., have profound effects in zero-IF receiver, which need not be considered in super-heterodyne receiver. Following a major study of flicker noise, possible solutions are given, especially those based on the passive mixer. Introduction Wireless communication becomes more and more important with the tendency of personal communication. Because wireless communication becomes digital, networking, small and intelligent, radio system will be changed from analog, digital to software. Accordingly, large translation will happen to the receiver architecture. [1] The receiver may usually be realized by super-heterodyne architecture. For super-heterodyne receiver, RF modulated signal can be shifted to easily processed IF and IF modulated signal can be handled: amplification, filtration and demodulation. Particular gain control, noise figure and narrow-band selection may be realized in super-heterodyne architecture, but there are some inherent disadvantages, such as complicated architecture, difficult coordination, bulk, and large power consumption. As a result, new receiver architecture appears. There are digital IF receiver, zero-IF receiver (also called direct-conversion receiver), etc., meeting the demands of high performance, low power consumption, agility and convenience. Because zero-IF receiver is similar to software-radio receiver in many ways, this paper presents flicker noise based on zero-IF receiver. -

Understanding Noise in the Signal Chain
Understanding Noise in the Signal Chain Introduction: What is the Signal Chain? A signal chain is any a series of signal-conditioning components that receive an input, passes the signal from component to component, and produces an output. Signal Chain Voltage Ref V in Amp ADC DSP DAC Amp Vout 2 Introduction: What is Noise? • Noise is any electrical phenomenon that is unwelcomed in the signal chain Signal Chain Ideal Signal Path, Gain (G) Voltage V V Ref int ext Internal Noise External Noise V in + Amp ADC DSP DAC Amp + G·(Vin + Vext) + Vint • Our focus is on the internal sources of noise – Noise in semiconductor devices in general – Noise in data converters in particular 3 Noise in Semiconductor Devices 1. How noise is specified a. Noise amplitude b. Noise spectral density 2. Types of noise a. White noise sources b. Pink noise sources 3. Reading noise specifications a. Time domain specs b. Frequency domain specs 4. Estimating noise amplitudes a. Creating a noise spectral density plot b. Finding the noise amplitude 4 Noise in Semiconductor Devices 1. How noise is specified a. Noise amplitude b. Noise spectral density 2. Types of noise a. White noise sources b. Pink noise sources 3. Reading noise specifications a. Time domain specs b. Frequency domain specs 4. Estimating noise amplitudes a. Creating a noise spectral density plot b. Finding the noise amplitude 5 Noise in Semiconductor Devices How Noise is Specified: Amplitude Noise Amplitude Semiconductor noise results from random processes and thus the instantaneous amplitude is unpredictable. Amplitude exhibits a Gaussian (Normal) distribution.