Rare Variants in Axonogenesis Genes Connect Three Families with Sound–Color Synesthesia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Neuronal Repellent SLIT2 Is a Target for Repression by EZH2 in Prostate Cancer
Oncogene (2010) 29, 5370–5380 & 2010 Macmillan Publishers Limited All rights reserved 0950-9232/10 www.nature.com/onc ORIGINAL ARTICLE The neuronal repellent SLIT2 is a target for repression by EZH2 in prostate cancer JYu1,2,3,4, Q Cao2,3,JYu2,LWu1, A Dallol5,JLi2, G Chen2, C Grasso2,3, X Cao2,3, RJ Lonigro2,4, S Varambally2,3, R Mehra2,3, N Palanisamy2,3,JYWu1,8, F Latif5 and AM Chinnaiyan2,3,4,6,7 1Division of Hematology/Oncology, Department of Medicine, Northwestern University, Robert H. Lurie Comprehensive Cancer Center, Chicago, IL, USA; 2Michigan Center for Translational Pathology, University of Michigan, Ann Arbor, MI, USA; 3Department of Pathology, University of Michigan, Ann Arbor, MI, USA; 4Comprehensive Cancer Center, University of Michigan, Ann Arbor, MI, USA; 5Department of Medical and Molecular Genetics, Institute of Biomedical Research, University of Birmingham, Edgbaston, UK; 6Howard Hughes Medical Institute, University of Michigan, Ann Arbor, MI, USA; 7Department of Urology, University of Michigan, Ann Arbor, MI, USA and 8Department of Neurology, Lurie Comprehensive Cancer Center, Center for Genetic Medicine, Northwestern University, Chicago, IL, USA The neuronal repellent SLIT2 is repressed in a number of also includes SLIT1 and SLIT3. The SLIT proteins are cancer types primarily through promoter hypermethyla- evolutionary conserved and contain an N-terminal signal tion. SLIT2, however, has not been studied in prostate peptide, four leucine-rich tandem repeats, seven or nine cancer. Through genome-wide location analysis we epidermal growth factor repeats, a laminin G domain and a identified SLIT2 as a target of polycomb group (PcG) C-terminal cysteine knot (Rothberg et al., 1988). -

A 1.37-Mb 12P11.22-P11.21 Deletion Coincident with a 367-Kb 22Q11.2
CORE Metadata, citation and similar papers at core.ac.uk Provided by Elsevier - Publisher Connector Taiwanese Journal of Obstetrics & Gynecology 53 (2014) 74e78 Contents lists available at ScienceDirect Taiwanese Journal of Obstetrics & Gynecology journal homepage: www.tjog-online.com Short Communication A 1.37-Mb 12p11.22ep11.21 deletion coincident with a 367-kb 22q11.2 duplication detected by array comparative genomic hybridization in an adolescent girl with autism and difficulty in self-care of menstruation Chih-Ping Chen a,b,c,d,e,f,*, Shuan-Pei Lin b,g,h,i, Schu-Rern Chern b, Peih-Shan Wu j, Jun-Wei Su a,k, Chen-Chi Lee a, Wayseen Wang b,l a Department of Obstetrics and Gynecology, Mackay Memorial Hospital, Taipei, Taiwan b Department of Medical Research, Mackay Memorial Hospital, Taipei, Taiwan c Department of Biotechnology, Asia University, Taichung, Taiwan d School of Chinese Medicine, College of Chinese Medicine, China Medical University, Taichung, Taiwan e Institute of Clinical and Community Health Nursing, National Yang-Ming University, Taipei, Taiwan f Department of Obstetrics and Gynecology, School of Medicine, National Yang-Ming University, Taipei, Taiwan g Department of Medicine, Mackay Medical College, New Taipei City, Taiwan h Department of Pediatrics, Mackay Memorial Hospital, Taipei, Taiwan i Mackay Junior College of Medicine, Nursing, and Management, Taipei, Taiwan j Gene Biodesign Co. Ltd, Taipei, Taiwan k Department of Obstetrics and Gynecology, China Medical University Hospital, Taichung, Taiwan l Department of Bioengineering, Tatung University, Taipei, Taiwan article info abstract Article history: Objective: To present an array comparative genomic hybridization (aCGH) characterization of a 12p11.22 Accepted 21 October 2013 ep11.21 microdeletion and 22q11.2 microduplication in an adolescent girl with autism, mental retar- dation, facial dysmorphism, microcephaly, behavior problems, and an apparently balanced reciprocal Keywords: translocation of t(8;12)(q24.3;p11.2). -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

DNA Replication Stress Response Involving PLK1, CDC6, POLQ
DNA replication stress response involving PLK1, CDC6, POLQ, RAD51 and CLASPIN upregulation prognoses the outcome of early/mid-stage non-small cell lung cancer patients C. Allera-Moreau, I. Rouquette, B. Lepage, N. Oumouhou, M. Walschaerts, E. Leconte, V. Schilling, K. Gordien, L. Brouchet, Mb Delisle, et al. To cite this version: C. Allera-Moreau, I. Rouquette, B. Lepage, N. Oumouhou, M. Walschaerts, et al.. DNA replica- tion stress response involving PLK1, CDC6, POLQ, RAD51 and CLASPIN upregulation prognoses the outcome of early/mid-stage non-small cell lung cancer patients. Oncogenesis, Nature Publishing Group: Open Access Journals - Option C, 2012, 1, pp.e30. 10.1038/oncsis.2012.29. hal-00817701 HAL Id: hal-00817701 https://hal.archives-ouvertes.fr/hal-00817701 Submitted on 9 Jun 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution - NonCommercial - NoDerivatives| 4.0 International License Citation: Oncogenesis (2012) 1, e30; doi:10.1038/oncsis.2012.29 & 2012 Macmillan Publishers Limited All rights reserved 2157-9024/12 www.nature.com/oncsis ORIGINAL ARTICLE DNA replication stress response involving PLK1, CDC6, POLQ, RAD51 and CLASPIN upregulation prognoses the outcome of early/mid-stage non-small cell lung cancer patients C Allera-Moreau1,2,7, I Rouquette2,7, B Lepage3, N Oumouhou3, M Walschaerts4, E Leconte5, V Schilling1, K Gordien2, L Brouchet2, MB Delisle1,2, J Mazieres1,2, JS Hoffmann1, P Pasero6 and C Cazaux1 Lung cancer is the leading cause of cancer deaths worldwide. -

Evolution of Language: Lessons from the Genome
Psychon Bull Rev DOI 10.3758/s13423-016-1112-8 BRIEF REPORT Evolution of language: Lessons from the genome Simon E. Fisher1,2 # The Author(s) 2016. This article is published with open access at Springerlink.com Abstract The post-genomic era is an exciting time for re- Keywords Genetics and genomics . Speech and language . searchers interested in the biology of speech and language. Evolution . Ancient DNA . Model systems Substantive advances in molecular methodologies have opened up entire vistas of investigation that were not previ- ously possible, or in some cases even imagined. Speculations Our speech and language capacities enable us to acquire vocab- concerning the origins of human cognitive traits are being ularies of many thousands of words, assemble them into a myr- transformed into empirically addressable questions, generat- iad of structured meaningful expressions, and convey thoughts ing specific hypotheses that can be explicitly tested using data to others by mapping meaning to sound, and back again. In the collected from both the natural world and experimental set- twenty-first century, we are witnessing dramatic advances in tings. In this article, I discuss a number of promising lines of deciphering the genetic architecture underlying these fascinat- research in this area. For example, the field has begun to ing aspects of the human condition. By directly borrowing identify genes implicated in speech and language skills, in- state-of-the-art gene mapping approaches from studies of typi- cluding not just disorders but also the normal range of abili- cal biomedical traits, and applying them to scientific studies of ties. Such genes provide powerful entry points for gaining language for the first time, it has become feasible to start tracing insights into neural bases and evolutionary origins, using so- out relevant genetic networks (Graham & Fisher, 2015). -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

Dynamic Landscape of Chromatin Accessibility and Transcriptomic
www.nature.com/scientificreports OPEN Dynamic landscape of chromatin accessibility and transcriptomic changes during diferentiation of human embryonic stem cells into dopaminergic neurons César Meléndez‑Ramírez1,2,6, Raquel Cuevas‑Diaz Duran3,6*, Tonatiuh Barrios‑García3, Mayela Giacoman‑Lozano3, Adolfo López‑Ornelas1,2,4, Jessica Herrera‑Gamboa3, Enrique Estudillo2, Ernesto Soto‑Reyes5, Iván Velasco1,2* & Víctor Treviño3* Chromatin architecture infuences transcription by modulating the physical access of regulatory factors to DNA, playing fundamental roles in cell identity. Studies on dopaminergic diferentiation have identifed coding genes, but the relationship with non‑coding genes or chromatin accessibility remains elusive. Using RNA‑Seq and ATAC‑Seq we profled diferentially expressed transcripts and open chromatin regions during early dopaminergic neuron diferentiation. Hierarchical clustering of diferentially expressed genes, resulted in 6 groups with unique characteristics. Surprisingly, the abundance of long non‑coding RNAs (lncRNAs) was high in the most downregulated transcripts, and depicted positive correlations with target mRNAs. We observed that open chromatin regions decrease upon diferentiation. Enrichment analyses of accessibility depict an association between open chromatin regions and specifc functional pathways and gene‑sets. A bioinformatic search for motifs allowed us to identify transcription factors and structural nuclear proteins that potentially regulate dopaminergic diferentiation. Interestingly, we also found changes in protein and mRNA abundance of the CCCTC‑binding factor, CTCF, which participates in genome organization and gene expression. Furthermore, assays demonstrated co‑localization of CTCF with Polycomb‑repressed chromatin marked by H3K27me3 in pluripotent cells, progressively decreasing in neural precursor cells and diferentiated neurons. Our work provides a unique resource of transcription factors and regulatory elements, potentially involved in the acquisition of human dopaminergic neuron cell identity. -
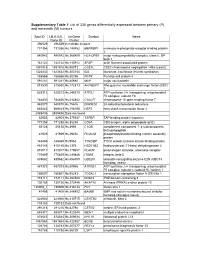
Supplementary Table 1 List of 335 Genes Differentially Expressed Between Primary (P) and Metastatic (M) Tumours
Supplementary Table 1 List of 335 genes differentially expressed between primary (P) and metastatic (M) tumours Spot ID I.M.A.G.E. UniGene Symbol Name Clone ID Cluster 296529 296529 In multiple clusters 731356 731356 Hs.140452 M6PRBP1 mannose-6-phosphate receptor binding protein 1 840942 840942 Hs.368409 HLA-DPB1 major histocompatibility complex, class II, DP beta 1 142122 142122 Hs.115912 AFAP actin filament associated protein 1891918 1891918 Hs.90073 CSE1L CSE1 chromosome segregation 1-like (yeast) 1323432 1323432 Hs.303154 IDS iduronate 2-sulfatase (Hunter syndrome) 788566 788566 Hs.80296 PCP4 Purkinje cell protein 4 591281 591281 Hs.80680 MVP major vault protein 815530 815530 Hs.172813 ARHGEF7 Rho guanine nucleotide exchange factor (GEF) 7 825312 825312 Hs.246310 ATP5J ATP synthase, H+ transporting, mitochondrial F0 complex, subunit F6 784830 784830 Hs.412842 C10orf7 chromosome 10 open reading frame 7 840878 840878 Hs.75616 DHCR24 24-dehydrocholesterol reductase 669443 669443 Hs.158195 HSF2 heat shock transcription factor 2 2485436 2485436 Data not found 82903 82903 Hs.370937 TAPBP TAP binding protein (tapasin) 771258 771258 Hs.85258 CD8A CD8 antigen, alpha polypeptide (p32) 85128 85128 Hs.8986 C1QB complement component 1, q subcomponent, beta polypeptide 41929 41929 Hs.39252 PICALM phosphatidylinositol binding clathrin assembly protein 148469 148469 Hs.9963 TYROBP TYRO protein tyrosine kinase binding protein 415145 415145 Hs.1376 HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 810017 810017 Hs.179657 PLAUR plasminogen activator, -

Variation in Protein Coding Genes Identifies Information
bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 1 1 2 3 4 5 Variation in protein coding genes identifies information flow as a contributor to 6 animal complexity 7 8 Jack Dean, Daniela Lopes Cardoso and Colin Sharpe* 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Institute of Biological and Biomedical Sciences 25 School of Biological Science 26 University of Portsmouth, 27 Portsmouth, UK 28 PO16 7YH 29 30 * Author for correspondence 31 [email protected] 32 33 Orcid numbers: 34 DLC: 0000-0003-2683-1745 35 CS: 0000-0002-5022-0840 36 37 38 39 40 41 42 43 44 45 46 47 48 49 Abstract bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 2 1 Across the metazoans there is a trend towards greater organismal complexity. How 2 complexity is generated, however, is uncertain. Since C.elegans and humans have 3 approximately the same number of genes, the explanation will depend on how genes are 4 used, rather than their absolute number. -

A Genome-Wide Association Study Identifies Four Novel Susceptibility Loci Underlying Inguinal Hernia
UCSF UC San Francisco Previously Published Works Title A genome-wide association study identifies four novel susceptibility loci underlying inguinal hernia. Permalink https://escholarship.org/uc/item/7g06z1k5 Journal Nature communications, 6(1) ISSN 2041-1723 Authors Jorgenson, Eric Makki, Nadja Shen, Ling et al. Publication Date 2015-12-21 DOI 10.1038/ncomms10130 Peer reviewed eScholarship.org Powered by the California Digital Library University of California ARTICLE Received 24 Aug 2015 | Accepted 6 Nov 2015 | Published 21 Dec 2015 DOI: 10.1038/ncomms10130 OPEN A genome-wide association study identifies four novel susceptibility loci underlying inguinal hernia Eric Jorgenson1,*, Nadja Makki2,3,*, Ling Shen1, David C. Chen4, Chao Tian5, Walter L. Eckalbar2,3, David Hinds5, Nadav Ahituv2,3 & Andrew Avins1 Inguinal hernia repair is one of the most commonly performed operations in the world, yet little is known about the genetic mechanisms that predispose individuals to develop inguinal hernias. We perform a genome-wide association analysis of surgically confirmed inguinal hernias in 72,805 subjects (5,295 cases and 67,510 controls) and confirm top associations in an independent cohort of 92,444 subjects with self-reported hernia repair surgeries (9,701 cases and 82,743 controls). We identify four novel inguinal hernia susceptibility loci in the regions of EFEMP1, WT1, EBF2 and ADAMTS6. Moreover, we observe expression of all four genes in mouse connective tissue and network analyses show an important role for two of these genes (EFEMP1 and WT1) in connective tissue maintenance/homoeostasis. Our findings provide insight into the aetiology of hernia development and highlight genetic pathways for studies of hernia development and its treatment. -

Transcriptomic and Proteomic Profiling Provides Insight Into
BASIC RESEARCH www.jasn.org Transcriptomic and Proteomic Profiling Provides Insight into Mesangial Cell Function in IgA Nephropathy † † ‡ Peidi Liu,* Emelie Lassén,* Viji Nair, Celine C. Berthier, Miyuki Suguro, Carina Sihlbom,§ † | † Matthias Kretzler, Christer Betsholtz, ¶ Börje Haraldsson,* Wenjun Ju, Kerstin Ebefors,* and Jenny Nyström* *Department of Physiology, Institute of Neuroscience and Physiology, §Proteomics Core Facility at University of Gothenburg, University of Gothenburg, Gothenburg, Sweden; †Division of Nephrology, Department of Internal Medicine and Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan; ‡Division of Molecular Medicine, Aichi Cancer Center Research Institute, Nagoya, Japan; |Department of Immunology, Genetics and Pathology, Uppsala University, Uppsala, Sweden; and ¶Integrated Cardio Metabolic Centre, Karolinska Institutet Novum, Huddinge, Sweden ABSTRACT IgA nephropathy (IgAN), the most common GN worldwide, is characterized by circulating galactose-deficient IgA (gd-IgA) that forms immune complexes. The immune complexes are deposited in the glomerular mesangium, leading to inflammation and loss of renal function, but the complete pathophysiology of the disease is not understood. Using an integrated global transcriptomic and proteomic profiling approach, we investigated the role of the mesangium in the onset and progression of IgAN. Global gene expression was investigated by microarray analysis of the glomerular compartment of renal biopsy specimens from patients with IgAN (n=19) and controls (n=22). Using curated glomerular cell type–specific genes from the published literature, we found differential expression of a much higher percentage of mesangial cell–positive standard genes than podocyte-positive standard genes in IgAN. Principal coordinate analysis of expression data revealed clear separation of patient and control samples on the basis of mesangial but not podocyte cell–positive standard genes. -

Appendix 2. Significantly Differentially Regulated Genes in Term Compared with Second Trimester Amniotic Fluid Supernatant
Appendix 2. Significantly Differentially Regulated Genes in Term Compared With Second Trimester Amniotic Fluid Supernatant Fold Change in term vs second trimester Amniotic Affymetrix Duplicate Fluid Probe ID probes Symbol Entrez Gene Name 1019.9 217059_at D MUC7 mucin 7, secreted 424.5 211735_x_at D SFTPC surfactant protein C 416.2 206835_at STATH statherin 363.4 214387_x_at D SFTPC surfactant protein C 295.5 205982_x_at D SFTPC surfactant protein C 288.7 1553454_at RPTN repetin solute carrier family 34 (sodium 251.3 204124_at SLC34A2 phosphate), member 2 238.9 206786_at HTN3 histatin 3 161.5 220191_at GKN1 gastrokine 1 152.7 223678_s_at D SFTPA2 surfactant protein A2 130.9 207430_s_at D MSMB microseminoprotein, beta- 99.0 214199_at SFTPD surfactant protein D major histocompatibility complex, class II, 96.5 210982_s_at D HLA-DRA DR alpha 96.5 221133_s_at D CLDN18 claudin 18 94.4 238222_at GKN2 gastrokine 2 93.7 1557961_s_at D LOC100127983 uncharacterized LOC100127983 93.1 229584_at LRRK2 leucine-rich repeat kinase 2 HOXD cluster antisense RNA 1 (non- 88.6 242042_s_at D HOXD-AS1 protein coding) 86.0 205569_at LAMP3 lysosomal-associated membrane protein 3 85.4 232698_at BPIFB2 BPI fold containing family B, member 2 84.4 205979_at SCGB2A1 secretoglobin, family 2A, member 1 84.3 230469_at RTKN2 rhotekin 2 82.2 204130_at HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 81.9 222242_s_at KLK5 kallikrein-related peptidase 5 77.0 237281_at AKAP14 A kinase (PRKA) anchor protein 14 76.7 1553602_at MUCL1 mucin-like 1 76.3 216359_at D MUC7 mucin 7,