Development and Validation of Acacia Koa and A. Koaia Nuclear Ssrs Using Illumina Sequencing
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Handbook Publication.Pub
Table of Contents Maui County’s Landscape and Gardening Handbook Xeriscaping in Maui County ................................................................. 1 Planning and Design................................................................................................................. 1 Hydro-zones.............................................................................................................................. 1 Plant Selection and the Maui jkCounty Planting Zones............................................................ 2 Soil Preparation ........................................................................................................................ 4 Mulching.................................................................................................................................... 5 Irrigation .................................................................................................................................... 5 Maintenance ............................................................................................................................. 7 Other Interesting Techniques for the Ambitious ..................................... 8 Xeriscape Ponds....................................................................................................................... 8 Aquaponics in the Backyard ..................................................................................................... 9 Water Polymer Crystals ........................................................................................................... -
Additional Information
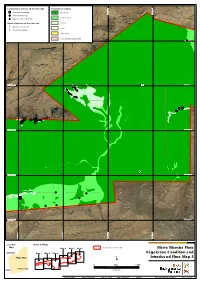
Current Survey Introduced Flora Records Vegetation Condition *Acetosa vesicaria Excellent 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 -

Tree Spacing Is Per the City and County of Honolulu, Department of Parks and Recreation, Division of Urban Forestry - Street Tree Specifications
Recommended Industry Standard Plant Spacing Guidelines TREES: Canopy Spread Street Tree No. Common Botanical Small Medium Large Height Spacing WRA Comments 1 `A`ali`i Dodonaea viscosa X < 30' 25 NL 2 `Ohai Ali`i Caesalpinia pulcherrima X < 20' 25 5 3 `Ohi`a Lehua Metrosideros polymorpha X 80' - 100' 40 NL 4 Alahe`e Psydrax odorata X 3' - 30' 25 NL 5 Autograph Clusia rosea X < 30' 60 5 6 Beach Heliotrope Tournefortia argentea X X 15' - 35' 40 -1 7 Breadfruit Artocarpus altilis X 60' N/A -12 8 Brown Pine Podocarpus elatus X 100' - 125' N/A -2 25' o.c. 9 Carrotwood Cupaniopsis anacardioides X 25' - 40' 40 9 10 Coral Erythrina crista-galli X < 30' 40 6 11 Crape Myrtle Lagerstroemia indica X X < 30' 25 6 12 False Olive Cassine orientalis X < 30' 40 -1 13 False Sandalwood (Naio) Myoporum sandwicense X 30' - 60' N/A NL 60' o.c. 14 Fern Podocarpus Afrocarpus gracilior X 20' - 40' 40 0 15 Geiger (Haole Kou) Cordia sebestena X < 30' N/A -1 40' o.c. 16 Geometry Bucida buceras X 45' - 60' 40 -3 17 Giant Crape Myrtle Lagerstroemia speciosa X 30' - 80' 60 -4 18 Gold tree Roseodendron donnell-smithii X 60' - 90' 85 -4 Handroanthus ochracea subsp. 19 Golden Trumpet neochrysantha X 40' - 60' 60 -3 20 Hala Pandanus tectorius X X < 35' N/A NL 25' o.c. 21 Hau Hibiscus tiliaceus X X < 35' N/A NL 40' o.c. 22 Hau (Variegated) Hibiscus tiliaceus forma X < 30' 25 NL 23 Ho`awa Pittosporum hosmeri X < 30' 25 NL 24 Hong Kong Orchid Bauhinia xblakeana X 25' - 35' 40 -7 Recommended Industry Standard Plant Spacing Guidelines TREES: Canopy Spread Street Tree No. -

Impacts of Alien Land Arthropods and Mollusks on Native Plants and Animals in Hawaii
7. IMPACTS OF ALIEN LAND ARTHROPODS AND MOLLUSKS ON NATIVE PLANTS AND ANIMALS IN HAWAIfI Francis G. Howarth ABSTRACT Over 2,000 alien arthropod species and about 30 alien non-marine mollusks are established in the wild in Hawai'i, While the data are too meager to assess fully the impacts of any of these organisms on the na- tive biota, the documentation suggests several areas of critical concern. Alien species feed directly on na- tive plants or their products, thus competing with na- tive herbivores and affecting host plants. Alien pred- ators and parasites critically reduce the populations of many native species and seriously deplete the food resources of native predators. Some immigrant species spread diseases that infect elements of the native bio- ta. Others are toxic to native predators. There is also competition for other resources, such as nesting and resting sites. Even apparently innocuous intro- duced species may provide food for alien predators, thus keeping predator populations high with an atten- dant greater impact on native prey. Control measures targeted at alien pests may be hazardous to natives. Mitigative measures must be based on sound research and firmer understanding of the complex interactions and dynamics of functioning ecosystems. Strict quarantine procedures are cost effective in preventing or delaying the establishment of potential pests. Strict control or fumigation is needed for nonessential importations (such as cow chips, Christmas trees, and flowers in bulk). Improved review of introductions for biological control is required in order to prevent repeating past mistakes. Biocontrol introductions must be used only for bona fide pests and used in native ecosystems only in special circumstances. -

Pu'u Wa'awa'a Biological Assessment
PU‘U WA‘AWA‘A BIOLOGICAL ASSESSMENT PU‘U WA‘AWA‘A, NORTH KONA, HAWAII Prepared by: Jon G. Giffin Forestry & Wildlife Manager August 2003 STATE OF HAWAII DEPARTMENT OF LAND AND NATURAL RESOURCES DIVISION OF FORESTRY AND WILDLIFE TABLE OF CONTENTS TITLE PAGE ................................................................................................................................. i TABLE OF CONTENTS ............................................................................................................. ii GENERAL SETTING...................................................................................................................1 Introduction..........................................................................................................................1 Land Use Practices...............................................................................................................1 Geology..................................................................................................................................3 Lava Flows............................................................................................................................5 Lava Tubes ...........................................................................................................................5 Cinder Cones ........................................................................................................................7 Soils .......................................................................................................................................9 -

Genetic Diversity and Differentiation of Invasive Acacia Longifolia in Portugal
Web Ecol., 18, 91–103, 2018 https://doi.org/10.5194/we-18-91-2018 © Author(s) 2018. This work is distributed under the Creative Commons Attribution 4.0 License. Genetic diversity and differentiation of invasive Acacia longifolia in Portugal Sara Vicente1,2, Cristina Máguas2, and Helena Trindade1 1Centro de Estudos do Ambiente e do Mar (CESAM), Centro de Biotecnologia Vegetal (CBV), Faculdade de Ciências da Universidade de Lisboa, Lisbon, 1749-016, Portugal 2Center for Ecology, Evolution and Environmental Changes (cE3c), Faculdade de Ciências da Universidade de Lisboa, Lisbon, 1749-016, Portugal Correspondence: Helena Trindade ([email protected]) Received: 30 January 2018 – Revised: 27 April 2018 – Accepted: 4 May 2018 – Published: 24 May 2018 Abstract. Acacia longifolia is an aggressive invader worldwide. This species was brought to Portugal by the forestry services and is now found throughout the country with very strong ecological and social impacts. Al- though several ecological and physiological studies have been carried out, molecular studies in this species are sparse. Particularly, genetic variability evaluation in invasive ranges clearly deserves more attention. The aim of this study was to evaluate the genetic diversity and the genetic differentiation among populations of the alien in- vasive A. longifolia under different Mediterranean conditions. For that we studied three sandy dune populations along the Portuguese coast: Osso da Baleia (mesomediterranean), Pinheiro da Cruz and Vila Nova de Milfontes (termomediterranean). All sampled acacia plants were located underneath a pine forest, with the exception of the latter case study, where we also sampled in a diversified habitat conditions associated with the margins of agriculture fields. -
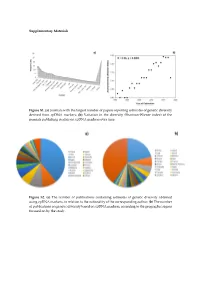
(A) Journals with the Largest Number of Papers Reporting Estimates Of
Supplementary Materials Figure S1. (a) Journals with the largest number of papers reporting estimates of genetic diversity derived from cpDNA markers; (b) Variation in the diversity (Shannon-Wiener index) of the journals publishing studies on cpDNA markers over time. Figure S2. (a) The number of publications containing estimates of genetic diversity obtained using cpDNA markers, in relation to the nationality of the corresponding author; (b) The number of publications on genetic diversity based on cpDNA markers, according to the geographic region focused on by the study. Figure S3. Classification of the angiosperm species investigated in the papers that analyzed genetic diversity using cpDNA markers: (a) Life mode; (b) Habitat specialization; (c) Geographic distribution; (d) Reproductive cycle; (e) Type of flower, and (f) Type of pollinator. Table S1. Plant species identified in the publications containing estimates of genetic diversity obtained from the use of cpDNA sequences as molecular markers. Group Family Species Algae Gigartinaceae Mazzaella laminarioides Angiospermae Typhaceae Typha laxmannii Angiospermae Typhaceae Typha orientalis Angiospermae Typhaceae Typha angustifolia Angiospermae Typhaceae Typha latifolia Angiospermae Araliaceae Eleutherococcus sessiliflowerus Angiospermae Polygonaceae Atraphaxis bracteata Angiospermae Plumbaginaceae Armeria pungens Angiospermae Aristolochiaceae Aristolochia kaempferi Angiospermae Polygonaceae Atraphaxis compacta Angiospermae Apocynaceae Lagochilus macrodontus Angiospermae Polygonaceae Atraphaxis -

List 01 Hawaiian Names 01 Plants
V\.{). 3 v BOTANICAL BULLETIN NO.2 JUNE. 1913 TERRITORY OF HAWAII BOARD OF AGRICULTURE AND FORESTRY List 01 Hawaiian Names 01 Plants BY JOSEPH F. ROCK Consulting Botanist, Board of Agriculture and Forestry HONOLULU: HAWAIIAN GAZETTE CO., LTD. 1913 ALPHABETICAL LIST OF HAWAIIAN NAMES OF PLANTS. The following list of Hawaiian plant-names has been compiled from various sources. Hillebrand in his valuable Flora of the Hawaiian Islands has given many Hawaiian names, especially of the more common species; these are incorporated in this list with a few corrections. Nearly all Hawaiian plant-names found in this list and not in Hillebrand's Flora were secured from Mr. Francis Gay of the Island of Kauai, an old resident in this Terri tory and well acquainted with its plants from a layman's stand point. It was the writer's privilege to camp with Mr. Gay in the mountains of Kauai collecting botanical material; for almost every species he could give the native name, which he had se cured in the early days from old and reliable natives. Mr. Gay had made spatter prints of many of the native plants in a large record book with their names and uses, as well as their symbolic meaning when occurring in mele (songs) or olioli (chants), at tached to them. For all this information the writer is indebted mainly to Mr. Francis Gay and also to Mr. Augustus F. Knudsen of the same Island. The writer also secured Hawaiian names from old na tives and Kahunas (priests) in the various islands of the group. -

The Following Tree Seedlings Are Available to Order from the State of Hawaii Division of Forestry and Wildlife, State Tree Nursery
The following tree seedlings are available to order from the State of Hawaii Division of Forestry and Wildlife, State Tree Nursery: Scientific Name: Common Name: Dibble/ Pot size: Acacia koa……………………… Koa……………………………….. Small Acacia koaia……………………... Koai’a……………………………. Small Araucaria columnaris…………….. Norfolk-island Pine……………… Small Cryptomeria japonica……………. Sugi Pine………………………… Small Cupressus lusitanica……………... Mexican Cypress………………… Small Cupressus macrocarpa…………… Monterey Cypress……………….. Small Cupressus simpervirens………….. Italian Cypress…………………… Medium Eucalyptus deglupta……………… Rainbow Bark……………………. Small Eucalyptus robusta……………….. Swamp Mahogany……………….. Small Metrosideros polymorpha……….. Ohia……………………………… Medium or 3” pot Pinus elliotii……………………… Slash Pine………………………... Small Pinus radiata……………………... Monterey Pine…………………… Small Podocarpus sp……………………. Podocarpus………………………. 3” pot Santalum sp……………………… Sandalwood……………………… Medium or 3” pot Tristania conferta………………… Brush Box………………………... Small Acacia koa (Koa): This large hardwood tree is endemic to the Hawaiian Islands. The tree has exceeded 100 ft in height with basal diameter far beyond 50 inches in old growth stands. The wood is prized for furniture and canoe works. This legume has pods with black seeds for reproduction. The wood has similar properties to that of black walnut. The yellow flowers are borne in dense round heads about 2@ in diameter. Tree growth is best above 800 ft; seems to grow best in the ‘Koa belt’ which is situated at an elevation range between 3,500 - 6,000 ft. It is often found in areas where there is fog in the late afternoons. It should be planted in well- drained fertile soils. Grazing animals relish the Koa foliage, so young seedlings should be protected Acacia koaia (Koaia): Related to the Koa, Koaia is native to Hawaii. The leaves and flowers are much the same as Koa. -

Farm and Forestry Production and Marketing Profile for Sandalwood (Santalum Species)
Specialty Crops for Pacific Island Agroforestry (http://agroforestry.net/scps) Farm and Forestry Production and Marketing Profile for Sandalwood (Santalum species) By Lex A.J. Thomson, John Doran, Danica Harbaugh, and Mark D. Merlin USES AND PRODUCTS apart from carving, sandalwood is rarely used in many of Wood from the Hawaiian and South Pacific sandalwoods these traditional ways nowadays because of its scarcity and traditionally had a diversity of uses such as carving, medi- economic value. cine, insect repellent when burnt (St. John 1947), and fuel Commercial exploitation of sandalwood commenced in the (Wagner 1986). The grated wood was used to a limited ex- Hawaiian and some South Pacific islands during the late 18th tent to scent coconut oil (for application to the hair and and early 19th centuries when the aromatic lower trunks and body) and cultural artifacts such as tapa cloth (Krauss 1993; rootstock of native Santalum species were harvested in great Kepler 1985). In Hawai‘i, sandalwoods were used to make quantity and shipped to China. There they were used to musical instruments such as the musical bow (‘ūkēkē) and make incense, fine furniture, and other products. The exten- to produce hoe (paddles) as part of traditional canoe con- sive and often exploitative sandalwood trade in Hawai‘i was struction (Buck 1964; Krauss 1993). The highest-value wood an early economic activity that adversely affected both the from the sandalwoods is used for carving (i.e., religious stat- natural environment and human health. Indeed, this activ- ues and objects, handicrafts, art, and decorative furniture). ity represented an early shift from subsistence to commer- For most purposes larger basal pieces and main roots are cial economy in Hawai‘i that was to have far-reaching and preferred due to their high oil content and better oil profiles, long-lasting effects in the islands (Shineberg 1967). -

Auwahi: Ethnobotany of a Hawaiian Dryland Forest
AUWAHI: ETHNOBOTANY OF A HAWAIIAN DRYLAND FOREST. A. C. Medeiros1, C.F. Davenport2, and C.G. Chimera1 1. U.S. Geological Survey, Biological Resources Division, Haleakala Field Station, P.O. Box 369, Makawao, HI 96768 2. Social Sciences Department, Maui Community College, 310 Ka’ahumanu Ave., Kahului, HI 96732 ABSTRACT Auwahi district on East Maui extends from sea level to about 6800 feet (1790 meters) elevation at the southwest rift of leeward Haleakal¯a volcano. In botanical references, Auwahi currently refers to a centrally located, fairly large (5400 acres) stand of diverse dry forest at 3000-5000 feet (915- 1525 meters) elevation surrounded by less diverse forest and more open-statured shrubland on lava. Auwahi contains high native tree diversity with 50 dryland species, many with extremely hard, durable, and heavy wood. To early Hawaiians, forests like Auwahi must have seemed an invaluable source of unique natural materials, especially the wide variety of woods for tool making for agriculture and fishing, canoe building, kapa making, and weapons. Of the 50 species of native trees at Auwahi, 19 species (38%) are known to have been used for medicine, 13 species (26%) for tool-making, 13 species (26%) for canoe building 13 species (26%) for house building, 8 species (16%) for tools for making kapa, 8 species (16%) for weapons 8 species (16%) for fishing, 8 species (16%) for dyes, and 7 species (14 %) for religious purposes. Other miscellaneous uses include edible fruits or seeds, bird lime, cordage, a fish narcotizing agent, firewood, a source of "fireworks", recreation, scenting agents, poi boards, and h¯olua sled construction. -

Hawaiian Santalum Species (Sandalwood)
April 2006 Species Profiles for Pacific Island Agroforestry ver. 4.1 www.traditionaltree.org Santalum ellipticum, S. freycinetianum, S. haleakalae, and S. paniculatum (Hawaiian sandalwood) Santalaceae (sandalwood family) ‘iliahialo‘e (S. ellipticum) ‘iliahi, ‘a‘ahi, ‘aoa, lā‘au ‘ala, wahie ‘ala (S. freycinetianum, S. haleakalae, and S. paniculatum) Mark D. Merlin, Lex A.J. Thomson, and Craig R. Elevitch IN BRIEF Distribution Hawaiian Islands, varies by species. Size Small shrubs or trees, typically 5–10 m photo: M. Merlin M. photo: (16–33 ft) or larger at maturity. Habitat Varies by species; typically xeric, sub humid, or humid tropics with a distinct dry season of 3–5 months. Vegetation Open, drier forests and wood lands. Soils All species require light to medium, well drained soils. Growth rate Slow to moderate, 0.3–0.7 m/yr (1–2.3 ft/yr). Main agroforestry uses Homegardens, mixed species forestry. Main uses Heartwood for crafts, essential oil extraction. Yields Heartwood in 30+ years. Intercropping Because sandalwood is para sitic and requires one or more host plants, intercropping is not only possible but neces sary. Santalum freycinetianum var. Invasive potential Sandalwood has a capac lanaiense, rare, nearly extinct. ity for invasiveness in disturbed areas, but this Photo taken near summit of is rarely considered a problem. Lāna‘ihale, Lāna‘i in 1978. INTRODUCTION SANDALWOOD TERMS Hawaiian sandalwood species are small trees that occur naturally in open, drier forest and woodland communities. Hemi-parasitic Describes a plant that photosynthe They are typically multistemmed and somewhat bushy, at sizes but derives water and some nutrients through at taining a height of 5–10 m (16–33 ft) or larger at maturity, taching to roots of other species.