The Role of Biotransformation in Drug Discovery and Development
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

WHO Pharmaceuticals Newsletters
2021 WHO Pharmaceuticals No.1 NEWSLETTER The WHO Pharmaceuticals Newsletter provides you with the latest information on the safety of medicines WHO Vision for Medicines Safety and legal actions taken by regulatory authorities around No country left behind: the world. It also provides signals based on information worldwide pharmacovigilance for safer medicines, safer patients derived from the WHO global database of individual case safety reports, VigiBase. In addition, this edition of the Newsletter includes a The aim of the Newsletter is to disseminate regulatory summary of discussions and key recommendations of information on the safety of Advisory Committee on Safety of Medicinal Products pharmaceutical products, (ACSoMP) Seventeenth meeting. based on communications received from our network of national pharmacovigilance centres and other sources such as specialized bulletins and journals, as well as partners in WHO. The information is produced in the form of résumés in English, full texts of which may be obtained on request from: Safety and Vigilance: Medicines, EMP-HIS, World Health Organization, 1211 Geneva 27, Switzerland, Contents E -mail address: [email protected] This Newsletter is also available at: Regulatory matters http://www.who.int/medicines Safety of medicines Signal Feature WHO Pharmaceuticals Newsletter No. 1, 2021 ISBN 978-92-4-002136-5 (electronic version) ISBN 978-92-4-002137-2 (print version) © World Health Organization 2021 Some rights reserved. This work is available under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. -

Clinical Pharmacology 1: Phase 1 Studies and Early Drug Development
Clinical Pharmacology 1: Phase 1 Studies and Early Drug Development Gerlie Gieser, Ph.D. Office of Clinical Pharmacology, Div. IV Objectives • Outline the Phase 1 studies conducted to characterize the Clinical Pharmacology of a drug; describe important design elements of and the information gained from these studies. • List the Clinical Pharmacology characteristics of an Ideal Drug • Describe how the Clinical Pharmacology information from Phase 1 can help design Phase 2/3 trials • Discuss the timing of Clinical Pharmacology studies during drug development, and provide examples of how the information generated could impact the overall clinical development plan and product labeling. Phase 1 of Drug Development CLINICAL DEVELOPMENT RESEARCH PRE POST AND CLINICAL APPROVAL 1 DISCOVERY DEVELOPMENT 2 3 PHASE e e e s s s a a a h h h P P P Clinical Pharmacology Studies Initial IND (first in human) NDA/BLA SUBMISSION Phase 1 – studies designed mainly to investigate the safety/tolerability (if possible, identify MTD), pharmacokinetics and pharmacodynamics of an investigational drug in humans Clinical Pharmacology • Study of the Pharmacokinetics (PK) and Pharmacodynamics (PD) of the drug in humans – PK: what the body does to the drug (Absorption, Distribution, Metabolism, Excretion) – PD: what the drug does to the body • PK and PD profiles of the drug are influenced by physicochemical properties of the drug, product/formulation, administration route, patient’s intrinsic and extrinsic factors (e.g., organ dysfunction, diseases, concomitant medications, -

DESCRIPTION CLINICAL PHARMACOLOGY Mechanism Of
NDA 20-844/ Topamav Sprinkle Capsules Approved Labeling Text Version: 10/26/98 DESCRIPTION Topiramate is a sulfamate-substituted monosaccharide that is intended for use as an antiepileptic drug. TOPAMAX@ (topiramate capsules) Sprinkle Capsules are available as I5 mg, 25 mg and 50mg sprinkle capsules for oral administration as whole capsules or for opening and sprinkling onto soft food. Topiramate is a white crystalline powder with a bitter taste. Topiramate is most soluble in alkaline solutions containing sodium hydroxide or sodium phosphate and hawng a pH of 9 to IO. It is freely soluble in acetone, chloroform, dimethylsulfoxide, and ethanol. The solubility in water is 9.8 mg/mL. Its saturated solution has a pH of 6.3. Topiramate has the molecular formula C,,H,,NO,S and a molecular weight of 339.37. Topiramate is designated chemically as 2,3:4,5-Di-O-isopropylidene-~- D-fructopyranose sulfamate and has the following structural formula: H3C CH3 TOPAMAX” (topiramate capsules) Sprinkle Capsules contain topiramate coated beads in a hard gelatin capsule. The inactive ingredients are: sugar spheres (sucrose and starch), povidone, cellulose acetate, gelatin, silicone dioxide, sodium lauryl sulfate, titanium dioxide, and black pharmaceutical ink. CLINICAL PHARMACOLOGY Mechanism of Action: The precise mechanism by which topiramate exerts its antiseizure effect is unknown; however, electrophysiological and biochemical studies of the effects of topiramate on cultured neurons have revealed three properties that may contribute to topiramate’s antiepileptic efficacy. First, action potentials elicited repetitively by a sustained depolarization of the neurons are blocked by topiramate in a time-dependent manner, suggestive of a state-dependent sodium channel blocking action. -

Plant Phenolics: Bioavailability As a Key Determinant of Their Potential Health-Promoting Applications
antioxidants Review Plant Phenolics: Bioavailability as a Key Determinant of Their Potential Health-Promoting Applications Patricia Cosme , Ana B. Rodríguez, Javier Espino * and María Garrido * Neuroimmunophysiology and Chrononutrition Research Group, Department of Physiology, Faculty of Science, University of Extremadura, 06006 Badajoz, Spain; [email protected] (P.C.); [email protected] (A.B.R.) * Correspondence: [email protected] (J.E.); [email protected] (M.G.); Tel.: +34-92-428-9796 (J.E. & M.G.) Received: 22 October 2020; Accepted: 7 December 2020; Published: 12 December 2020 Abstract: Phenolic compounds are secondary metabolites widely spread throughout the plant kingdom that can be categorized as flavonoids and non-flavonoids. Interest in phenolic compounds has dramatically increased during the last decade due to their biological effects and promising therapeutic applications. In this review, we discuss the importance of phenolic compounds’ bioavailability to accomplish their physiological functions, and highlight main factors affecting such parameter throughout metabolism of phenolics, from absorption to excretion. Besides, we give an updated overview of the health benefits of phenolic compounds, which are mainly linked to both their direct (e.g., free-radical scavenging ability) and indirect (e.g., by stimulating activity of antioxidant enzymes) antioxidant properties. Such antioxidant actions reportedly help them to prevent chronic and oxidative stress-related disorders such as cancer, cardiovascular and neurodegenerative diseases, among others. Last, we comment on development of cutting-edge delivery systems intended to improve bioavailability and enhance stability of phenolic compounds in the human body. Keywords: antioxidant activity; bioavailability; flavonoids; health benefits; phenolic compounds 1. Introduction Phenolic compounds are secondary metabolites widely spread throughout the plant kingdom with around 8000 different phenolic structures [1]. -

Moving Beyond Single Gene-Drug Pairs in Clinical Pharmacogenomics Testing
Moving Beyond Single Gene-Drug Pairs in Clinical Pharmacogenomics Testing Yuan Ji, PhD, DABCP, FACMG Gwendolyn McMillin, PhD, DABCC Learning Objectives • Describe the strengths and limitations of pharmacogenomic testing. • List examples of single gene-drug associations with the strongest levels of evidence for clinical implementation. • Discuss cautions when considering the use of multi-gene drug associations to inform drug therapy decisions. 2 Disclosure • None 3 Outline • Singe gene-drug based pharmacogenomics (PGx) testing – An introduction – Evidence and examples – Considerations for developing and evaluating clinical PGx laboratory developed tests (LDTs) • Multi-gene PGx panels – Evidence and examples – PROs and CONs for utilizing PGx panels – Considerations for successful PGx implementation 4 Single Gene-Drug Based PGx Testing Yuan Ji, PhD, DABCP, FACMG Medical Director of Genomics and Genetics; PGx, ARUP Laboratories Associate Professor (Clinical) of Pathology, University of Utah Medications: Myths and Facts • are~30% peoplenot take“one at least-size one medication fits all” within a 30-day period • Most medications cause adverse drug events (ADEs) • Some medications like antibiotics may do more harm than good • CDC statistics: – ~ 200,000 ADEs-related ER visits in pediatric population (17 years or younger) – ~ 450,000 ADEs-related ER visits in older adults (65 years or older) – Medications, e.g., anticoagulant warfarin • Many ADEs are preventable by closely supervising of dosing, blood tests (therapeutic drug monitoring, TDM), -

Metabolism of Non-Growing Cells and Their Application in Biotransformation
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 17 July 2020 doi:10.20944/preprints202007.0402.v1 Metabolism of non-growing cells and their application in biotransformation Wenfa Ng Department of Chemical and Biomolecular Engineering, National University of Singapore, Email: [email protected] Abstract Growing cells is the typical mode of operation in many aspects of biotechnology and metabolic engineering. This comes about due to cell growth processes creating a driving force that pull metabolic flux along different metabolic pathways, that indirectly help move substrate to product. But, there is an alternative mode of operation that uses resting (non-growing) cells to achieve similar or even higher productivities. In general, resting cells are provided with carbon substrates for biocatalytic reactions but starved of nitrogen or phosphorus. Such resting cells have been usefully employed in many forms of biocatalysis and biotransformation, with or without cofactor regeneration. However, much remains unknown about the transcriptome and metabolome of resting cells in biotransformation settings. This short writeup provides the backdrop of resting cells in biocatalysis, documents their use in biotransformation with application examples, and identifies research gaps that could be filled with contemporary RNA- seq and mass spectrometry proteomics technology. Overall, utility of resting cells in biocatalysis and the extant knowledge gap in their fundamental physiology are highlighted in this resource. Keywords: resting cells, biocatalysis, cofactor regeneration, transcriptome, biotransformations, Subject areas: biochemistry, biotechnology, bioinformatics, systems biology, molecular biology, Non-growing (resting) cells in biotransformation Biotransformations sought the use of enzymes and whole cells for the conversion of substrates into desired products. Typically, whole cell biocatalysts are used given problems with instability and purification of pure enzymes. -
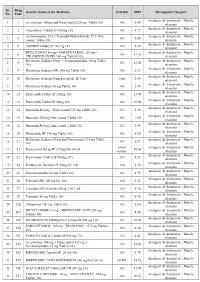
PMBJP Product.Pdf
Sr. Drug Generic Name of the Medicine Unit Size MRP Therapeutic Category No. Code Analgesic & Antipyretic / Muscle 1 1 Aceclofenac 100mg and Paracetamol 325 mg Tablet 10's 10's 8.00 relaxants Analgesic & Antipyretic / Muscle 2 2 Aceclofenac Tablets IP 100mg 10's 10's 4.37 relaxants Acetaminophen 325 + Tramadol Hydrochloride 37.5 film Analgesic & Antipyretic / Muscle 3 4 10's 8.00 coated Tablet 10's relaxants Analgesic & Antipyretic / Muscle 4 5 ASPIRIN Tablets IP 150 mg 14's 14's 2.70 relaxants DICLOFENAC 50 mg+ PARACETAMOL 325 mg+ Analgesic & Antipyretic / Muscle 5 6 10's 11.30 CHLORZOXAZONE 500 mg Tablets 10's relaxants Diclofenac Sodium 50mg + Serratiopeptidase 10mg Tablet Analgesic & Antipyretic / Muscle 6 8 10's 12.00 10's relaxants Analgesic & Antipyretic / Muscle 7 9 Diclofenac Sodium (SR) 100 mg Tablet 10's 10's 6.12 relaxants Analgesic & Antipyretic / Muscle 8 10 Diclofenac Sodium 25mg per ml Inj. IP 3 ml 3 ml 2.00 relaxants Analgesic & Antipyretic / Muscle 9 11 Diclofenac Sodium 50 mg Tablet 10's 10's 2.90 relaxants Analgesic & Antipyretic / Muscle 10 12 Etoricoxilb Tablets IP 120mg 10's 10's 33.00 relaxants Analgesic & Antipyretic / Muscle 11 13 Etoricoxilb Tablets IP 90mg 10's 10's 25.00 relaxants Analgesic & Antipyretic / Muscle 12 14 Ibuprofen 400 mg + Paracetamol 325 mg Tablet 10's 15's 5.50 relaxants Analgesic & Antipyretic / Muscle 13 15 Ibuprofen 200 mg film coated Tablet 10's 10's 1.80 relaxants Analgesic & Antipyretic / Muscle 14 16 Ibuprofen 400 mg film coated Tablet 10's 15's 3.50 relaxants Analgesic & Antipyretic -

Smitha MS, Singh S, Singh R. Microbial Biotransformation: a Process for Chemical Alterations
Journal of Bacteriology & Mycology: Open Access Mini Review Open Access Microbial biotransformation: a process for chemical alterations Abstract Volume 4 Issue 2 - 2017 Biotransformation is a process by which organic compounds are transformed from one Smitha MS, Singh S, Singh R form to another to reduce the persistence and toxicity of the chemical compounds. This Amity University Uttar Pradesh, India process is aided by major range of microorganisms and their products such as bacteria, fungi and enzymes. Biotransformations can also be used to synthesize compounds Correspondence: Singh R, Additional Director, Amity Institute or materials, if synthetic approaches are challenging. Natural transformation process of Microbial Biotechnology, Amity University, Sector 125, Noida, is slow, nonspecific and less productive. Microbial biotransformations or microbial UP, India, Tel +911204392900, Email [email protected] biotechnology are gaining importance and extensively utilized to generate metabolites in bulk amounts with more specificity. This review was conceived to assess the Received: November 21, 2016 | Published: February 21, 2017 impact of microbial biotransformation of steroids, antibiotics, various pollutants and xenobiotic compounds. Keywords: microbial biotransformation; bioconversion; metabolism; steroid; bacteria, fungi Introduction the metabolism of compounds using animal models.9 The microbial biotransformation phenomenon is then commonly employed Biotransformations are structural modifications in a chemical in comparing metabolic -

Toxicological Review of Phenol
EPA/635/R-02/006 TOXICOLOGICAL REVIEW OF Phenol (CAS No. 108-95-2) In Support of Summary Information on the Integrated Risk Information System (IRIS) September 2002 U.S. Environmental Protection Agency Washington D.C. DISCLAIMER Mention of trade names or commercial products does not constitute endorsement or recommendation for use. Note: This document may undergo revisions in the future. The most up-to-date version will be made available electronically via the IRIS Home Page at http://www.epa.gov/iris. CONTENTS - TOXICOLOGICAL REVIEW FOR PHENOL (CAS No. 108-95-2) Foreword................................................................... vi Authors, Contributors and Reviewers.......................................... vii 1. INTRODUCTION ......................................................1 2. CHEMICAL AND PHYSICAL INFORMATION RELEVANT TO ASSESSMENTS........................................................2 3. TOXICOKINETICS RELEVANT TO ASSESSMENTS ......................5 3.1 Absorption ......................................................5 3.2 Distribution.....................................................10 3.3 Metabolism.....................................................12 3.4 Excretion .......................................................23 4. HAZARD IDENTIFICATION ...........................................24 4.1 Studies in Humans - Epidemiology, Case Reports, Clinical Controls .....25 4.1.1 Oral .....................................................25 4.1.2 Inhalation ................................................27 4.2 -

Biosynthesis of New Alpha-Bisabolol Derivatives Through a Synthetic Biology Approach Arthur Sarrade-Loucheur
Biosynthesis of new alpha-bisabolol derivatives through a synthetic biology approach Arthur Sarrade-Loucheur To cite this version: Arthur Sarrade-Loucheur. Biosynthesis of new alpha-bisabolol derivatives through a synthetic biology approach. Biochemistry, Molecular Biology. INSA de Toulouse, 2020. English. NNT : 2020ISAT0003. tel-02976811 HAL Id: tel-02976811 https://tel.archives-ouvertes.fr/tel-02976811 Submitted on 23 Oct 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. THÈSE En vue de l’obtention du DOCTORAT DE L’UNIVERSITÉ DE TOULOUSE Délivré par l'Institut National des Sciences Appliquées de Toulouse Présentée et soutenue par Arthur SARRADE-LOUCHEUR Le 30 juin 2020 Biosynthèse de nouveaux dérivés de l'α-bisabolol par une approche de biologie synthèse Ecole doctorale : SEVAB - Sciences Ecologiques, Vétérinaires, Agronomiques et Bioingenieries Spécialité : Ingénieries microbienne et enzymatique Unité de recherche : TBI - Toulouse Biotechnology Institute, Bio & Chemical Engineering Thèse dirigée par Gilles TRUAN et Magali REMAUD-SIMEON Jury -

Immediately Dangerous to Life Or Health (IDLH) Value Profile: Butane
Butane CAS® No. 106-97-8 DEPARTMENT OF HEALTH AND HUMAN SERVICES Center for Disease Control and Prevention National Institute of Occupational Safety and Health This page intentionally left blank. Immediately Dangerous to Life or Health (IDLH) Value Profile Butane [CAS® No. 106-97-8] H3C CH3 DEPARTMENT OF HEALTH AND HUMAN SERVICES Centers for Disease Control and Prevention National Institute for Occupational Safety and Health This document is in the public domain and may be freely copied or reprinted. Disclaimer Mention of any company or product does not constitute endorsement by the National Institute for Occupational Safety and Health (NIOSH). In addition, citations to websites external to NIOSH do not constitute NIOSH endorsement of the sponsoring organizations or their programs or products. Furthermore, NIOSH is not responsible for the content of these websites. Ordering Information To receive this document or information about other occupational safety and health topics, contact NIOSH: Telephone: 1-800-CDC-INFO (1-800-232-4636) TTY: 1-888-232-6348 E-mail: [email protected] or visit the NIOSH ebsite at: www.cdc.gov/niosh For a monthly update on news at NIOSH, subscribe to NIOSH eNews by visiting www.cdc.gov/niosh/eNews. Suggested Citation NIOSH [2016]. Immediately dangerous to life or health (IDLH) value profile: butane. By Dotson GS, Maier A, Parker A, Haber L. Cincinnati, OH: US Department of Health and Human Services, Centers for Disease Control and Prevention, National Institute for Occupa- tional Safety and Health, DHHS (NIOSH) Publication 2016-174. DHHS (NIOSH) Publication No. 2016-174 September 2016 ii IDLH Value Profile for Butane Foreword Chemicals are a ubiquitous component of the modern workplace. -

Microbial Transformation of Xenobiotics for Environmental Bioremediation
African Journal of Biotechnology Vol. 8 (22), pp. 6016-6027, 16 November, 2009 Available online at http://www.academicjournals.org/AJB ISSN 1684–5315 © 2009 Academic Journals Review Microbial transformation of xenobiotics for environmental bioremediation Shelly Sinha1, Pritam Chattopadhyay2, Ieshita Pan1, Sandipan Chatterjee1, Pompee Chanda1, 1 1 1 Debashis Bandyopadhyay , Kamala Das and Sukanta K. Sen * 1Microbiology Division, Department of Botany, Visva-Bharati, Santiniketan, 731 235, India. 2Plant Biotechnology Division, Department of Botany, Visva-Bharati, Santiniketan, 731 235, India. Accepted 4 September, 2009 The accumulation of recalcitrant xenobiotic compounds is due to continuous efflux from population and industrial inputs that have created a serious impact on the pristine nature of our environment. Apart from this, these compounds are mostly carcinogenic, posing health hazards which persist over a long period of time. Metabolic pathways and specific operon systems have been found in diverse but limited groups of microbes that are responsible for the transformation of xenobiotic compounds. Distinct catabolic genes are either present on mobile genetic elements, such as transposons and plasmids, or the chromosome itself that facilitates horizontal gene transfer and enhances the rapid microbial transformation of toxic xenobiotic compounds. Biotransformation of xenobiotic compounds in natural environment has been studied to understand the microbial ecology, physiology and evolution for their potential in bioremediation. Recent advance in the molecular techniques including DNA fingerprinting, microarrays and metagenomics is being used to augment the transformation of xenobiotic compounds. The present day understandings of aerobic, anaerobic and reductive biotransformation by co-metabolic processes and an overview of latest developments in monitoring the catabolic genes of xenobiotic-degrading bacteria are discussed elaborately in this work.