1 Population Genomics on the Origin of Lactase Persistence in Europe And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Cases for Evolution Education Question Guide By: Merle K Heidemann, Jim J Smith, Alexa Warwick, Louise Mead, Peter JT White
A Cases for Evolution Education Question Guide By: Merle K Heidemann, Jim J Smith, Alexa Warwick, Louise Mead, Peter JT White The Case of Lactase Persistence Evolution in Humans This Evo-Ed case consists of three modules that support the teaching and learning of biology in the framework of evolution of lactase persistence in humans. Together, the modules present evidence that evolution occurs because of: 1) competition for resources and differential reproductive success in populations 2) heritable genetic variation and resulting differences in gene expression. The following activities are designed to guide students’ learning as they engage in the modules of this case. They can also be used as learning objectives. That is, “students will be able to” accomplish each of these as objectives. The modules and activities are presented in the order in which they appear in the case and can be used as in-class activities, homework and/or formative assessments. The background information on this case, and accompanying slides can be found at: www.evo-ed.org/Pages/Lactase The Cell Biology of Lactase Persistence 1) Describe how the components of lactose become available for absorption into the blood stream. 2) Conduct research to determine if other sugars are digested in the same way as lactose. 3) Additionally, conduct research on how proteins and fats are digested in the small intestine. 4) Explain how the structure of enterocytes enables their function of absorbing digested molecules. 5) Describe the result, both at the cellular level and at the whole organism level, of lactose not being digested. 6) Research the cellular structure of a neuron (nerve cell - see clam toxin case). -

The Origins of Lactase Persistence and Ongoing Convergent Evolution
THE ORIGINS OF LACTASE PERSISTENCE AND ONGOING CONVERGENT EVOLUTION by BETH A. KELLER B.S. University of Phoenix, 2003 A thesis submitted in partial fulfillment of the requirements for the degree of Master of Arts in the Department of Anthropology in the College of Sciences at the University of Central Florida Orlando, Florida Spring Term 2011 ABSTRACT As a primary factor in human evolution, natural selection is an important component of genetic research. Studies of lactase persistence suggest that positive selection has played a powerful role in the adaptation to a lifelong consumption of fresh milk. Using multiple research studies of lactase persistence and suspected corresponding single nucleotide genetic polymorphisms, this study combines data sources to determine whether evidence exists for natural selection of a specific cytosine-to-thymine genetic mutation located 13,910 base pairs (T-13910) upstream from the lactase gene. This polymorphism has potential to be a causal element for lactase persistence, and data suggest that natural selection has played a role in the rising frequency and distribution of this allele, if only in some regions. European and neighboring regions appear to have the highest frequencies with little or no frequency in Asia, Africa and Indonesia; however the presence of lactase persistence in those areas suggests convergent evolution may be occurring on a phenotypic level. To examine this possibility several other identified polymorphisms in the same region as the T-13910 will be included in this study. ii Dedicated to my wonderful family whose encouragement made all the difference. iii ACKNOWLEDGMENTS I would like to thank my committee members, Dr. -

Lactase Persistence, Dietary Intake of Milk, and the Risk for Prostate Cancer in Sweden and Finland
956 Lactase Persistence, Dietary Intake of Milk, and the Risk for Prostate Cancer in Sweden and Finland Suvi Torniainen,1 Maria Hedelin,3 Ville Autio,4 Heli Rasinpera¨,1 Katarina Augustsson Ba¨lter,3 A˚ sa Klint,3 Rino Bellocco,3,8 Fredrik Wiklund,3 Pa¨r Stattin,9 Tarja Ikonen,6 Teuvo L.J. Tammela,7 Johanna Schleutker,5,6 Henrik Gro¨nberg,3 and Irma Ja¨rvela¨1,2 1Department of Medical Genetics, University of Helsinki; 2Laboratory of Molecular Genetics, Helsinki University Central Hospital, Helsinki, Finland; 3Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Stockholm, Sweden; 4Research Unit, Tampere University Hospital; 5Department of Clinical Chemistry, Tampere University Hospital; 6Laboratory of Cancer Genetics, Institute of Medical Technology and 7Department of Urology, Tampere University Hospital and Medical School, University of Tampere, Tampere, Finland; 8Department of Statistics, University of Milano-Bicocca, Milan, Italy; and 9Department of Surgery and Perioperative Sciences, Urology and Andrology, Umea˚University Hospital, Umea˚, Sweden Abstract Prostate carcinoma is the most common cancer in men. Its controls (Cancer Prostate in Sweden I study) using a primary pathogenesis is mostly unknown. Dairy products questionnaire. Only the consumption of low-fat milk was containing lactose have been suggested to be risk factors for found to be associated with increased risk of prostate cancer prostate cancer. Digestion of lactose is dependent on lactase [odds ratio (OR), 1.73; 95% confidence interval (95% CI), 1.16- activity in the intestinal wall. A single nucleotide polymor- 2.39]. A statistically significantly higher (P < 0.01) lactose phism C to T residing 13,910 bp upstream of the lactase gene intake was observed among subjects with high lactase activity has been shown to associate with the developmental down- (C/T and T/T genotypes) compared with those with low lactase regulation of lactase activity underlying persistence/non- activity (C/C genotype). -

Insights from Central Asian Herders and Ancient
Why and when was lactase persistence selected for? Insights from Central Asian herders and ancient DNA Laure Ségurel, Perle Guarino-Vignon, Nina Marchi, Sophie Lafosse, Romain Laurent, Céline Bon, Alexandre Fabre, Tatyana Hegay, Evelyne Heyer To cite this version: Laure Ségurel, Perle Guarino-Vignon, Nina Marchi, Sophie Lafosse, Romain Laurent, et al.. Why and when was lactase persistence selected for? Insights from Central Asian herders and ancient DNA. PLoS Biology, Public Library of Science, 2020, 18 (6), pp.e3000742. 10.1371/journal.pbio.3000742. inserm-02908479 HAL Id: inserm-02908479 https://www.hal.inserm.fr/inserm-02908479 Submitted on 29 Jul 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. PLOS BIOLOGY UNSOLVED MYSTERY Why and when was lactase persistence selected for? Insights from Central Asian herders and ancient DNA 1 1 1¤ 1 Laure SegurelID *, Perle Guarino-VignonID , Nina MarchiID , Sophie Lafosse , 1 1 2,3 4 Romain LaurentID , CeÂline BonID , Alexandre FabreID , Tatyana HegayID , 1 Evelyne HeyerID 1 Eco-anthropologie, MuseÂum national -

Temple Frieze from Iraq 2500 BCE
Outline Evolution of Lactase Persistence I History of dairying I Lactose and lactase Alan R. Rogers I Dairying without lactase I Evidence for natural selection March 27, 2016 I When did lactase persistence evolve in Europe? Domesticated Cattle Prehistory of dairying I Earliest fossils: 8000 BP (Near East) ∼ I 7500 BP: Milk residues in pottery around Sea of Marmara, I Maybe 9000 BP (Sahara) Turkey. I Uses I 6800 BP: up Danube River I Draft animal I 6000 BP: Britain, Scandinavia, central Europe I Meat I Blood (like the Masai) I 5300 BP: East into steppes, but not past Ural Mountains. I Sour milk Temple Frieze from Iraq 2500 BCE 39 Milking (right) and milk-processing (left) depicted on a temple frieze, c. 2500 BC, from Tell al-’Ubaid, Iraq. Dairy farming was an easy way to ensure a constant input of Delacroix, Ovid among the Scythians protein into the diet. Pastoralists adapted to it. Natural selection favoured those whose ability to digest milk was not switched off after weaning. Several different genes for lactose tolerance have been discovered. As we shall see in Chapter 9, their spread across Europe is a key piece of evidence for the important role of migrations later than those of the early farmers. Farming had gradually spread west across Anatolia. After a sparse sprinkling of pre-ceramic sites, Fikir Tepe and similar settlements were established by the Sea of Marmara between 2 7000 BC and 6400 BC. Cattle were not predominant in the early stages. Domestic sheep and goats seem to have spread across Anatolia first, followed by cattle in about 6500 BC.3 The high rainfall and greener grazing of the coastal lowland favoured cattle-keeping. -

Lactase Persistence and Lactase Non-Persistence to My Family and Friends Örebro Studies in Medicine 48
Lactase Persistence and Lactase Non-Persistence To my family and friends Örebro Studies in Medicine 48 Ricardo Almon Lactase Persistence and Lactase Non-Persistence Prevalence, influence on body fat, body height, and relation to the metabolic syndrome © Ricardo Almon, 2010 Title: Lactase Persistence and Lactase Non-Persistence. Prevalence, influence on body fat, body height, and relation to the metabolic syndrome. Publisher: Örebro University 2010 www.publications.oru.se [email protected] Printer: Intellecta Infolog, Kållered 11/2010 issn 1652-4063 isbn 978-91-7668-767-3 Abstract A single nucleotide polymorphism (SNP) in chromosome 2 gives humans the capability to continue digesting lactose after weaning. Among individuals of European descent it is the SNP (LCT-13910 C>T), which is located 14 kb upstream from the start of transcription of the lactase gene (LCT). A C to T mutation enables the continued production of the enzyme lactase throughout life, and thereby the digestion of lactose throughout life without symptoms of lactose intolerance. The trait is called lactase persistence (LP). LP is an autosomal dominant trait. In contrast, lactase non-persistent (LNP) individuals show a decline of lactase production after weaning. LNP individuals habitually show symptoms of lactose intolerance after consumption of milk and some milk products. Using the LCT -13910 C>T SNP we reassessed the prevalence of LP/LNP in Sweden. We increased the accuracy of our estimates by using Hardy-Weinberg’s formula for allelic frequencies. We found a prevalence of 14% for LNP. This is about 5.5 fold higher than the prevalence formerly assumed for Sweden (range: 1-5%). -

Evolutionary Genetics of Lactase Persistence in Eurasian Human Populations
Allma Mater Studiiorum – Uniiversiità dii Bollogna DOTTORATO DI RICERCA IN Biodiversità ed Evoluzione Ciclo XXVI Settore Concorsuale di afferenza: 05/B1 Zoologia e Antropologia Settore Scientifico disciplinare: BIO/08 Antropologia EVOLUTIONARY GENETICS OF LACTASE PERSISTENCE IN EURASIAN HUMAN POPULATIONS Presentata da: Sara De Fanti Coordinatore Dottorato Relatore Prof. Barbara Mantovani Prof.ssa Donata Luiselli Correlatore Dott. Marco Sazzini Esame finale anno 2014 Table of contents 1. Introduction 5 1.1 Human Genome Variability 5 1.1.1 Human Genome Variability 5 1.1.2 Genetic markers 11 1.1.3 Linkage Disequilibrium and Haplotypes 14 1.1.4 Processes Shaping Diversity 16 1.2 Lactase persistence 19 1.2.1 The LCT gene 19 1.2.2 Lactase Persistence 20 1.2.3 Identification of lactase phenotypes 22 1.2.4 Hypotheses for lactase persistence 23 1.2.5 Polymorphisms involved in lactase persistence phenotypes 25 1.2.6 Lactase persistence: archaeological evidence 30 2. Aim of The Study 35 3. Materials and Methods 39 3.1 Population Samples 39 3.1.1 Italian samples 39 3.1.2 Samples from Arabian Peninsula 39 3.2 SNPs Selection 40 3.3 SNPs Genotyping and Quality Control 42 3.3.1 PicoGreen Quantification 42 3.3.2 Multiplex PCR and SNPs Design 44 3.3.3 The Sequenom MassARRAY iPLEX Platform 45 3.3.4 Quality Control 47 1 3.4 Statistical Analyses 48 3.4.1 Linkage Disequilibrium Analysis 48 3.4.2 Allele Frequency Analysis 49 3.4.3 Haplotypes Reconstruction 50 3.4.4 Summary and population differentiation statistics 50 3.4.5 LD-based SNP pruning 51 3.4.5.1 Multivariate analyses 52 3.4.6 Analysis of the Molecular Variance 53 3.4.7 Phylogenetic Methods and Networks 53 3.4.8 Correlation Analysis 56 4. -
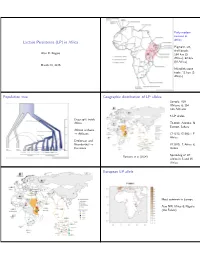
Lactase Persistence (LP) in Africa Population Tree Geographic
Early modern humans in Africa Lactase Persistence (LP) in Africa Pigment, art, shell beads: Alan R. Rogers 164 kya (S Africa); 82 kya (N Africa) March 30, 2016 Microlith stone tools: 71 kya (S Africa) Population tree Geographic distribution of LP alleles Sample: 819 Africans & 154 non-Africans 5 LP alleles Deep split inside Africa T13910, A22018: N Europe, Sahara African archaics Africans C14010, G13907: E → Africa Denisovan and Neanderthal G13915: E Africa & → Eurasians Arabia Ranciaro et al (2014) Sprinkling of LP alleles in S and W Africa. European LP allele Most common in Europe Also NW Africa & Nigeria (the Fulani) Arabian LP allele E African LP alleles Most common in Arabia Also E and C Africa Evidence for selection at C14010 Evidence for selection at C14010 Predominantly E African allele Predominantly E African allele in Kenya & Tanzania Extend Haplotype Homozygosity in Kenya & Tanzania (EHH) extends for 2 Mb (mega ∼ bases) in derived allele. EHH extends only a little way in ancestral allele. Strong evidence for recent selection. Evidence for selection at T13910 Evidence for selection at T13910 Predominantly European allele Extend Haplotype Homozygosity Predominantly European allele among Fulani (EHH) extends for 1.7 Mb in among Fulani derived allele. Only about 1 Mb in Europe. EHH extends only a little way in ancestral allele. Strong evidence for recent selection. LD scan showing two SNPs associated with LP Soft sweep in E Africa? E Africa Hard selective sweep Begins from a single copy of an advantageous mutation. Easy to detect with LD. Soft selective sweep Begins from multiple copies, possibly of multiple alleles with similar advantageous functions. -

The Genetic Variation of Lactase Persistence Alleles in Northeast Africa
bioRxiv preprint doi: https://doi.org/10.1101/2020.04.23.057356; this version posted April 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. i “output” — 2020/4/23 — 10:39 — page 1 — #1 i i i The genetic variation of lactase persistence alleles in northeast Africa ,1 2 1,3 1,4 Nina Hollfelder,⇤ Hiba Babiker, Lena Graneh¨all, Carina M Schlebusch, and Mattias ,1,4 Jakobsson⇤ 1Human Evolution, Department of Organismal Biology, Uppsala University, Uppsala, Sweden 2Department of Linguistic and Cultural Evolution, Max Planck Institute for the Science of Human History, Jena, Germany 3Institute for Mummy Studies, Eurac Research, Bolzano, Italy 4SciLifeLab, Uppsala University, Uppsala, Sweden ⇤Corresponding author: E-mail: [email protected], [email protected]. Associate Editor: Abstract Lactase persistence (LP) is a well-studied example of a Mendelian trait under selection in some human groups due to gene-culture co-evolution. We investigated the frequencies of genetic variants linked to LP in Sudanese and South Sudanese populations. These populations have diverse subsistence patterns, and some are dependent on milk to various extents, not only from cows, but also from other livestock such as camels and goats. We sequenced a 316bp region involved in regulating the expression of the LCT gene on chromosome 2, which encompasses five polymorphisms that have been associated with LP. Pastoralist populations showed a higher frequency of LP-associated alleles compared to non-pastoralist groups, hinting at positive selection also in northeast African pastoralists. -

Absence of the Lactase-Persistence-Associated Allele in Early Neolithic Europeans
Absence of the lactase-persistence-associated allele in early Neolithic Europeans J. Burger†‡, M. Kirchner†, B. Bramanti†, W. Haak†, and M. G. Thomas§ †Johannes Gutenberg University, Institute of Anthropology, Saarstrasse 21, D-55099 Mainz, Germany; and §Department of Biology, University College London, Wolfson House, 4 Stephenson Way, London NW1 2HE, United Kingdom Edited by Walter Bodmer, Cancer Research UK, Oxford, United Kingdom, and approved December 27, 2006 (received for review September 4, 2006) Lactase persistence (LP), the dominant Mendelian trait conferring would have provided a selective advantage in the absence of a the ability to digest the milk sugar lactose in adults, has risen to supply of fresh milk, and because of observed correlations high frequency in central and northern Europeans in the last 20,000 between the frequency of LP and the extent of traditional years. This trait is likely to have conferred a selective advantage in reliance on animal milk, the culture-historical hypothesis has individuals who consume appreciable amounts of unfermented been proposed (8–12). Under this model, LP was driven from milk. Some have argued for the ‘‘culture-historical hypothesis,’’ very rare cases of preadaptation to appreciable frequencies only whereby LP alleles were rare until the advent of dairying early in after the cultural practice of dairying arose. However, an op- the Neolithic but then rose rapidly in frequency under natural posing view, the reverse cause hypothesis, has also been pro- selection. Others favor the ‘‘reverse cause hypothesis,’’ whereby posed (8, 13, 14). According to this model, human populations dairying was adopted in populations with preadaptive high LP were already differentiated with regard to LP frequency before allele frequencies. -

Natural Selection and Adaptation
The Making of the Fittest: Got Lactase? NaturalThe Making Selection of the and Fittest: Adaptation The Co-evolution of Genes and Culture TEACHER MATERIALS Natural Selection and Adaptation IN-DEPTH FILM GUIDE DESCRIPTION Human babies drink milk; it’s the food especially produced for and given to them by their mothers. But when they grow into adults, most humans lose the ability to digest lactose, the sugar in milk. Why then do adults in some cultures think nothing of drinking a glass of cow’s milk? In this film, human geneticist Dr. Spencer Wells tracks down the genetic changes associated with the ability to digest lactose as an adult—a trait called lactase persistence—tracing the origin of the trait to pastoralist cultures that lived less than 10,000 years ago. Combining genetics, chemistry, anthropology, and biology, this story provides one of the most compelling and fascinating examples of gene-culture co-evolution. KEY CONCEPTS A. Humans, like all species, evolve and adapt to the environment through natural selection. Lactase persistence is an example of a human adaptation that arose within the last 10,000 years in response to a cultural change. B. Mutations occur at random; for evolution to occur there must be selection for or against the traits affected by those mutations. C. Both the physical and cultural environment can affect selective pressures. The practice of dairying provided an environment in which lactase persistence was advantageous. D. Different mutations can produce the same phenotype. Scientists have identified distinct mutations among northern Europeans and the Maasai people of eastern Africa that resulted in lactase persistence. -
Convergent Adaptation of Human Lactase Persistence in Africa and Europe
Europe PMC Funders Group Author Manuscript Nat Genet. Author manuscript; available in PMC 2009 April 23. Published in final edited form as: Nat Genet. 2007 January ; 39(1): 31–40. doi:10.1038/ng1946. Europe PMC Funders Author Manuscripts Convergent adaptation of human lactase persistence in Africa and Europe Sarah A Tishkoff1,9, Floyd A Reed1,9, Alessia Ranciaro1,2, Benjamin F Voight3, Courtney C Babbitt4, Jesse S Silverman4, Kweli Powell1, Holly M Mortensen1, Jibril B Hirbo1, Maha Osman5, Muntaser Ibrahim5, Sabah A Omar6, Godfrey Lema7, Thomas B Nyambo7, Jilur Ghori8, Suzannah Bumpstead8, Jonathan K Pritchard3, Gregory A Wray4, and Panos Deloukas8 1Department of Biology, University of Maryland, College Park, Maryland 20742, USA 2Department of Biology, University of Ferrara, 44100 Ferrara, Italy 3Department of Human Genetics, University of Chicago, Chicago, Illinois 60637, USA 4Institute for Genome Sciences & Policy and Department of Biology, Duke University, Durham, North Carolina 27708, USA 5Department of Molecular Biology, Institute of Endemic Diseases, University of Khartoum, 15-13 Khartoum, Sudan 6Kenya Medical Research Institute, Centre for Biotechnology Research and Development, 54840-00200 Nairobi, Kenya 7Department of Biochemistry, Muhimbili University College of Health Sciences, Dar es Salaam, Tanzania Europe PMC Funders Author Manuscripts 8Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge CB10 1SA, UK Abstract A SNP in the gene encoding lactase (LCT) (C/T-13910) is associated with the ability to digest milk as adults (lactase persistence) in Europeans, but the genetic basis of lactase persistence in Africans was previously unknown. We conducted a genotype-phenotype association study in 470 Correspondence should be addressed to S.A.T.