Study Protocol 1002-046 Amendment 1, 10 April 2017
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Homozygous Familial Hypercholesterolemia - Juxtapid® (Lomitapide Capsules)
Cigna National Formulary Coverage Policy Prior Authorization Homozygous Familial Hypercholesterolemia - Juxtapid® (lomitapide capsules) Table of Contents Product Identifer(s) National Formulary Medical Necessity ................ 1 52036 Conditions Not Covered....................................... 3 Background .......................................................... 3 References .......................................................... 4 Revision History ................................................... 4 INSTRUCTIONS FOR USE The following Coverage Policy applies to health benefit plans administered by Cigna Companies. Certain Cigna Companies and/or lines of business only provide utilization review services to clients and do not make coverage determinations. References to standard benefit plan language and coverage determinations do not apply to those clients. Coverage Policies are intended to provide guidance in interpreting certain standard benefit plans administered by Cigna Companies. Please note, the terms of a customer’s particular benefit plan document [Group Service Agreement, Evidence of Coverage, Certificate of Coverage, Summary Plan Description (SPD) or similar plan document] may differ significantly from the standard benefit plans upon which these Coverage Policies are based. For example, a customer’s benefit plan document may contain a specific exclusion related to a topic addressed in a Coverage Policy. In the event of a conflict, a customer’s benefit plan document always supersedes the information in the Coverage Policies. In the absence of a controlling federal or state coverage mandate, benefits are ultimately determined by the terms of the applicable benefit plan document. Coverage determinations in each specific instance require consideration of 1) the terms of the applicable benefit plan document in effect on the date of service; 2) any applicable laws/regulations; 3) any relevant collateral source materials including Coverage Policies and; 4) the specific facts of the particular situation. -

Effects of Pitavastatin, Atorvastatin, and Rosuvastatin on the Risk Of
biomedicines Article Effects of Pitavastatin, Atorvastatin, and Rosuvastatin on the Risk of New-Onset Diabetes Mellitus: A Single-Center Cohort Study Wei-Ting Liu 1, Chin Lin 2,3,4, Min-Chien Tsai 5, Cheng-Chung Cheng 6, Sy-Jou Chen 7,8, Jun-Ting Liou 6 , Wei-Shiang Lin 6, Shu-Meng Cheng 6, Chin-Sheng Lin 6,* and Tien-Ping Tsao 6,9,* 1 Department of Internal Medicine, Tri-Service General Hospital, National Defense Medical Center, Taipei 11490, Taiwan; [email protected] 2 School of Public Health, National Defense Medical Center, Taipei 11490, Taiwan; [email protected] 3 School of Medicine, National Defense Medical Center, Taipei 11490, Taiwan 4 Graduate Institute of Life Sciences, National Defense Medical Center, Taipei 11490, Taiwan, 5 Department of Physiology and Biophysics, Graduate Institute of Physiology, National Defense Medical Center, Taipei 11490, Taiwan; [email protected] 6 Division of Cardiology, Department of Internal Medicine, Tri-Service General Hospital, National Defense Medical Center, Taipei 11490, Taiwan; [email protected] (C.-C.C.); [email protected] (J.-T.L.); [email protected] (W.-S.L.); [email protected] (S.-M.C.) 7 Department of Emergency Medicine, Tri-Service General Hospital, National Defense Medical Center, Taipei 11490, Taiwan; [email protected] 8 Graduate Institute of Injury Prevention and Control, College of Public Health and Nutrition, Taipei Medical University, Taipei 11031, Taiwan 9 Division of Cardiology, Cheng Hsin General Hospital, Taipei 11220, Taiwan * Correspondence: [email protected] (C.-S.L.); [email protected] (T.-P.T.); Tel.: +886-2-6601-2656 (C.-S.L.); +886-2-2826-4400 (T.-P.T.) Received: 25 October 2020; Accepted: 11 November 2020; Published: 13 November 2020 Abstract: Statins constitute the mainstay treatment for atherosclerotic cardiovascular disease, which is associated with the risk of new-onset diabetes mellitus (NODM). -

Ezetimibe: a Novel Selective Cholesterol Absorption Inhibitor by Michele Koder, Pharm.D
OREGON DUR BOARD NEWSLETTER A N E VIDENCE B ASED D RUG T HERAPY R ESOURCE COPYRIGHT 2003 OREGON STATE UNIVERSITY. ALL RIGHTS RESERVED Volume 5, Issue 2 Also available on the web and via e-mail list-serve at February 2003 http://pharmacy.orst.edu/drug_policy/newsletter_email.html Ezetimibe: A novel selective cholesterol absorption inhibitor By Michele Koder, Pharm.D. , OSU College of Pharmacy Ezetimibe (Zetia) is a novel selective cholesterol absorption inhibitor that was approved by the FDA in October 2002. Unlike statins (HMG-CoA reductase inhibitors) and bile acid sequestrants, ezetimibe does not inhibit hepatic cholesterol synthesis or increase bile acid secretion. In contrast, ezetimibe selectively inhibits the uptake of dietary cholesterol from enterocytes in the brush border of the intestinal lumen resulting in a decrease in the delivery of dietary cholesterol to the liver and a subsequent decrease in hepatic cholesterol stores and increased cholesterol clearance from the blood.1 Ezetimibe’s unique action has generated interest in its use in combination with other cholesterol-lowering agents. It is indicated for the treatment of primary hypercholesterolemia as monotherapy and in combination with a statin. Ezetimibe is also approved for homozygous familial hypercholesterolemia and homozygous sitosterolemia. TABLE 1: EZETIMIBE CLINICAL TRIAL SUMMARY Study / Design Population Treatment % Change LDL % Change HDL % Change TG Bays et al3 N=432 EZ 5 mg -15.7 +2.9 MC, R, DB, PC LDL 130-250mg/dl EZ 10 mg -18.5 +3.5 NS 12 wk; Phase II TG -

Nustendi, INN-Bempedoic Acid, Ezetimibe
Summary of risk management plan for Nustendi (Bempedoic acid/Ezetimibe) This is a summary of the risk management plan (RMP) for Nustendi. The RMP details important risks of Nustendi, how these risks can be minimized, and how more information will be obtained about Nustendi's risks and uncertainties (missing information). Nustendi's summary of product characteristics (SmPC) and its package leaflet give essential information to healthcare professionals and patients on how Nustendi should be used. This summary of the RMP for Nustendi should be read in the context of all this information, including the assessment report of the evaluation and its plain-language summary, all which is part of the European Public Assessment Report (EPAR). Important new concerns or changes to the current ones will be included in updates of Nustendi's RMP. I. The Medicine and What It Is Used For Nustendi is authorized for treatment of primary hypercholesterolemia in adults, as an adjunct to diet (see SmPC for the full indication). It contains bempedoic acid as the active substance and it is given by mouth. Further information about the evaluation of Nustendi’s benefits can be found in Nustendi’s EPAR, including in its plain-language summary, available on the EMA website, under the medicine’s webpage https://www.ema.europa.eu/en/medicines/human/EPAR/nustendi II. Risks Associated With the Medicine and Activities to Minimize or Further Characterize the Risks Important risks of Nustendi, together with measures to minimize such risks and the proposed studies for learning -

Bempedoic Acid
Drug Therapy Guidelines Bempedoic Acid (Nexletol [bempedoic acid], Nexlizet [bempedoic acid, ezetimibe]) Applicable Medical Benefit Effective: 5/28/2021 Pharmacy- Formulary 1 x Next Review: 3/23 Pharmacy- Formulary 2 x Date of Origin: 4/23/20 Pharmacy- Formulary 3/Exclusive x Review Dates: 3/20, 3/21 Pharmacy- Formulary 4/AON x I. Medication Description Bempedoic acid is an adenosine triphosphate-citrate lyase (ACL) inhibitor that lowers low-density lipoprotein cholesterol (LDL-C) through inhibition of cholesterol synthesis in the liver. Bempedoic acid, an inactive prodrug, and its active metabolite, ESP15228, require coenzyme A (CoA) activation within the liver by very-long-chain acyl CoA synthetase-1 (ACSVL1) to form bempedoic acid-CoA (ETC-1002-CoA) and ESP15228-CoA, respectively. ACSVL1 is primarily expressed in the liver and is absent in adipose tissue and muscles. ACL is an important enzyme that links carbohydrate metabolism to the pathways for cholesterol and fatty synthesis within the liver. Specifically, ACL catalyzes the cleavage of citrate to acetyl-CoA and oxaloacetate within the cholesterol synthesis pathway and is located upstream from HMG-CoA reductase. Both oxaloacetate and acetyl-CoA are important substrates in the synthesis of cholesterol and fatty acids. Thus, inhibition of ACL by bempedoic acid decreases cholesterol and fatty acid synthesis resulting in the upregulation of LDL-C receptors, increased uptake of LDL-C by the liver and reduced blood LDL-C levels. Ezetimibe reduces blood cholesterol by inhibiting the absorption of cholesterol by the small intestine. The molecular target of ezetimibe has been shown to be the sterol transporter, Niemann-Pick C1-Like 1 (NPC1L1), which is involved in the intestinal uptake of cholesterol and phytosterols. -

Format (Sample) Dissertation
Providers’ Knowledge, Perceptions and Views of Prescribing Long-Acting Reversible Contraception to Adolescents in a Southwest Community Health Center Item Type text; Electronic Dissertation Authors Schafer, Stephanie Lynne Publisher The University of Arizona. Rights Copyright © is held by the author. Digital access to this material is made possible by the University Libraries, University of Arizona. Further transmission, reproduction or presentation (such as public display or performance) of protected items is prohibited except with permission of the author. Download date 02/10/2021 06:38:23 Link to Item http://hdl.handle.net/10150/626699 PROVIDERS’ KNOWLEDGE, PERCEPTIONS AND VIEWS OF PRESCRIBING LONG-ACTING REVERSIBLE CONTRACEPTION TO ADOLESCENTS IN A SOUTHWEST COMMUNITY HEALTH CENTER by Stephanie Lynne Schafer ________________________ Copyright © Stephanie Lynne Schafer 2017 A DNP Project Submitted to the Faculty of the COLLEGE OF NURSING In Partial Fulfillment of the Requirements For the Degree of DOCTOR OF NURSING PRACTICE In the Graduate College THE UNIVERSITY OF ARIZONA 2 0 1 7 2 3 STATEMENT BY AUTHOR This DNP Project has been submitted in partial fulfillment of requirements for an advanced degree at The University of Arizona and is deposited in the University Library to be made available to borrowers under rules of the Library. Brief quotations from this DNP Project are allowable without special permission, provided that accurate acknowledgment of source is made. Requests for permission for extended quotation from or reproduction of this manuscript in whole or in part may be granted by the head of the major department or the Dean of the Graduate College when in his or her judgment the proposed use of the material is in the interests of scholarship. -

Assessing the Availability, Service Quality, and Price of Essential Medicines In
Assessing the Availability, Service Quality, and Price of Essential Medicines in Private Pharmacies in Afghanistan Norio Kasahara A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Washington 2015 Reading Committee: Louis P. Garrison, Jr., Chair Joseph B. Babigumira Andy Stergachis Program Authorized to Offer Degree: Pharmaceutical Outcomes Research and Policy ©Copyright 2015 Norio Kasahara ii Table of Contents Abstract ................................................................................................................................................................................... ................................................................................................ ................................................................................................ .................................................................................. ............... vvv Acknowledgements ................................................................................................................................................................................... ................................................................................................ ................................................................................. ............ viiviivii Summary ................................................................................................................................................................................... ............................................................................................... -
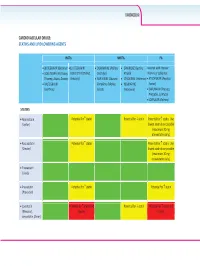
Statins and Lipid Lowering Agents
CARDIOVASCULAR Z/Ks^h>ZZh'^͗ ^dd/E^E>/W/>KtZ/E''Ed^ /E^d/Ɛ EEZd/Ɛ W/Ɛ x/d'Zs/Z ;ŝŬƚĂƌǀLJͿ x>s/d'Zs/Zͬ x KZs/Z/E ;WŝĨĞůƚƌŽ͕ x &s/ZE ;^ƵƐƚŝǀĂ͕ ŽŽƐƚĞĚǁŝƚŚƌŝƚŽŶĂǀŝƌ xK>hd'Zs/Z ;dŝǀŝĐĂLJ͕ K//^dd ;^ƚƌŝďŝůĚ͕ ĞůƐƚƌŝŐŽͿ ƚƌŝƉůĂͿ ;EŽƌǀŝƌͿŽƌĐŽďŝĐŝƐƚĂƚ dƌŝƵŵĞƋ͕:ƵůƵĐĂ͕ŽǀĂƚŽͿ 'ĞŶǀŽLJĂͿ x Z/>W/s/Z/E ;ĚƵƌĂŶƚ͕ x dZs/Z/E ;/ŶƚĞůĞŶĐĞͿ xdEs/Z ;ZĞLJĂƚĂnj͕ xZ>d'Zs/Z ŽŵƉůĞƌĂ͕KĚĞĨƐĞLJ͕ x Es/ZW/E ǀŽƚĂnjͿ ;/ƐĞŶƚƌĞƐƐͿ :ƵůƵĐĂͿ ;sŝƌĂŵƵŶĞͿ xZhEs/Z ;WƌĞnjŝƐƚĂ͕ WƌĞnjĐŽďŝdž͕^LJŵƚƵnjĂͿ x>KW/Es/Z ;<ĂůĞƚƌĂͿ ^dd/E^ xƚŽƌǀĂƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ nƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ pƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ nƐƚĂƚŝŶ͘hƐĞ ;>ŝƉŝƚŽƌͿ ůŽǁĞƐƚƐƚĂƚŝŶĚŽƐĞƉŽƐƐŝďůĞ ;ŵĂdžŝŵƵŵϮϬŵŐ ĂƚŽƌǀĂƐƚĂƚŝŶĚĂŝůLJͿ͘ xZŽƐƵǀĂƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ nƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ nƐƚĂƚŝŶ͘hƐĞ ;ƌĞƐƚŽƌͿ ůŽǁĞƐƚƐƚĂƚŝŶĚŽƐĞƉŽƐƐŝďůĞ ;ŵĂdžŝŵƵŵϭϬŵŐ ƌŽƐƵǀĂƐƚĂƚŝŶĚĂŝůLJͿ͘ xWŝƚĂǀĂƐƚĂƚŝŶ ;>ŝǀĂůŽͿ xWƌĂǀĂƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ nƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ nƐƚĂƚŝŶ ;WƌĂǀĂĐŚŽůͿ x>ŽǀĂƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ nƐƚĂƚŝŶĂŶĚ WŽƚĞŶƚŝĂůĨŽƌ pƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ nƐƚĂƚŝŶĂŶĚ ;DĞǀĂĐŽƌͿ͕ ƚŽdžŝĐŝƚLJ ƚŽdžŝĐŝƚLJ ƐŝŵǀĂƐƚĂƚŝŶ ;ŽĐŽƌͿ CARDIOVASCULAR INSTIs NNRTIs PIs xBICTEGRAVIR (Biktarvy) xELVITEGRAVIR/ x DORAVIRINE (Pifeltro, x EFAVIRENZ (Sustiva, Boosted with ritonavir xDOLUTEGRAVIR (Tivicay, COBICISTAT (Stribild, Delstrigo) Atripla) (Norvir) or cobicistat Triumeq, Juluca) Genvoya) x RILPIVIRINE (Edurant, x ETRAVIRINE (Intelence) xATAZANAVIR (Reyataz, xRALTEGRAVIR Complera, Odefsey, x NEVIRAPINE Evotaz) (Isentress) Juluca) (Viramune) xDARUNAVIR (Prezista, Prezcobix, Symtuza) xLOPINAVIR (Kaletra) FIBRATES xFenofibrate, bezafibrate, gemfibrozil CHOLESTEROL ABSORPTION INHIBITOR xEzetimibe -

Role of Colesevelam in Combination Lipid-Lowering Therapy
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Springer - Publisher Connector Am J Cardiovasc Drugs (2013) 13:315–323 DOI 10.1007/s40256-013-0037-0 REVIEW ARTICLE Role of Colesevelam in Combination Lipid-Lowering Therapy Michael R. Jones • Oliseyenum M. Nwose Published online: 3 August 2013 Ó The Author(s) 2013. This article is published with open access at Springerlink.com Abstract Hyperlipidemia is associated with an increased ezetimibe, statin plus niacin, or statin plus ezetimibe) and risk of cardiovascular events; reducing low-density lipo- high-sensitivity C-reactive protein (with statins), and protein cholesterol (LDL-C), the primary target for cho- increases in apo A-I (with statins, ezetimibe, or statins plus lesterol-lowering therapy, lowers the risk for such events. niacin). Triglyceride levels remained relatively unchanged Although bile acid sequestrants were the first class of drugs when colesevelam was combined with statins, fibrates, to show a mortality benefit related to LDL-C lowering, ezetimibe, or statin plus ezetimibe, and decreased with the statins are now considered first-line pharmacological ther- triple combination of colesevelam, statin, and niacin. apy for reducing LDL-C levels because of their potency Colesevelam offset the negative glycemic effects of statins and their remarkable record of successful outcomes studies. and niacin in subjects with insulin resistance or impaired Nevertheless, a substantial proportion of patients do not glucose tolerance. Colesevelam was generally well toler- achieve LDL-C goals with statin monotherapy. In addition, ated when added to other lipid-lowering therapies in clin- because of adverse effects (primarily myopathy), some ical trials, with gastrointestinal effects such as constipation patients may be unwilling to use or unable to tolerate statin being the predominant adverse events. -

Cholestagel®) Combination and Monotherapy for Familial Hypercholesterolaemia
New Medicine Recommendation Colesevelam (Cholestagel®) Combination and monotherapy for familial hypercholesterolaemia Recommendation: Black - NOT recommended for use by the NHS in Lancashire. Colesevelam (Cholestagel®) is NOT recommended as combination or monotherapy for familial hypercholesterolaemia. Clinical evidence indicates that the drug is poorly tolerated and the trials were weak in significant areas such as the inclusion of small numbers of patients, only demonstrating surrogate endpoints or being conducted using nonstandard co-administered drugs. There are similar, established drugs for the treatment of the condition for which there were no head to head trials with Colesevalam therefore relative clinical efficacy cannot be directly demonstrated. Summary of supporting evidence: Primary dyslipidaemias, including familial hypercholesterolaemia (FH), are lipid disorders that are genetic in origin. FH is the most common inherited lipid metabolism disorder in its heterozygous form. In the UK heterozygous FH affects 1 in 500 of the UK population. [1] Statin therapy is the first-line treatment for patients with the vast majority of dyslipidaemias. NICE CG 71 recommends the prescribing of a high-intensity statin at the maximum licensed dose or at the maximum tolerated dose to achieve a reduction in LDL cholesterol concentration of greater than 50% from baseline. [2] Colesevelam is a lipid-lowering polymer that binds bile acids in the intestine, impeding their reabsorption. [3] Colesevelam is a third-line agent, behind statins and ezetimibe. [2] The main body of evidence for the use of colesevelam in familial hypercholesterolaemia comes from the European Public Assessment Reports (EPAR). Study GTC-48-301 was a randomised, double-blind, parallel design, placebo-controlled pivotal phase III dose-response study. -

FIELD Study Revealed Fenofibrate Reduced Need for Laser Treatment for Diabetic Retinopathy by Anthony C
Supplement to Supported by an unrestricted educational grant from Abbott Laboratories March/April 2008 FIELD Study Revealed Fenofibrate Reduced Need for Laser Treatment for Diabetic Retinopathy By Anthony C. Keech, MBBS, Msc Epid, FRANZCS, FRACP; and Paul Mitchell, MBBS(Hons), MD, PhD, FRANZCO, FRACS, FRCOphth, FAFPHM This agent’s mechanism of benefit in diabetic retinopathy appears to go beyond its effects on lipid concentration or blood pressure, and this potential mechanism of action operates even when glycemic control and blood pressure levels are within goal. ABSTRACT icant relative reduction was seen of almost one-third in PURPOSE the rate of first laser application for retinopathy after The FIELD (Fenofibrate Intervention and Event an average treatment duration of 5 years with fenofi- Lowering in Diabetes) study sought to investigate brate 200 mg/day. whether long-term lipid-lowering therapy with fenofi- In this report, we detail the effects of fenofibrate brate would reduce macro- and microvascular compli- administration on ophthalmic microvascular compli- cations among patients with type 2 diabetes. We previ- cations and attempt to clarify some of the underlying ously reported that in type 2 diabetes patients with pathologies being addressed among patients undergo- adequate glycemic and blood pressure control, a signif- ing laser treatment. Jointly sponsored by The Dulaney Foundation and Retina Today MARCH/APRIL 2008 I SUPPLEMENT TO RETINA TODAY I 1 FIELD Study Revealed Fenofibrate Reduced Need for Laser Treatment for Diabetic Retinopathy Jointly sponsored by The Dulaney Foundation and Retina Today. Release date: April 2008. Expiration date: April 2009. This continuing medical education activity is supported by an unrestricted educational grant from Abbott Laboratories. -

Zetia® (Ezetimibe) Tablets
29480958T REV 14 ZETIA® (EZETIMIBE) TABLETS DESCRIPTION ZETIA (ezetimibe) is in a class of lipid-lowering compounds that selectively inhibits the intestinal absorption of cholesterol and related phytosterols. The chemical name of ezetimibe is 1-(4-fluorophenyl)- 3(R)-[3-(4-fluorophenyl)-3(S)-hydroxypropyl]-4(S)-(4-hydroxyphenyl)-2-azetidinone. The empirical formula is C24H21F2NO3. Its molecular weight is 409.4 and its structural formula is: OH OH S SR N F F O Ezetimibe is a white, crystalline powder that is freely to very soluble in ethanol, methanol, and acetone and practically insoluble in water. Ezetimibe has a melting point of about 163°C and is stable at ambient temperature. ZETIA is available as a tablet for oral administration containing 10 mg of ezetimibe and the following inactive ingredients: croscarmellose sodium NF, lactose monohydrate NF, magnesium stearate NF, microcrystalline cellulose NF, povidone USP, and sodium lauryl sulfate NF. CLINICAL PHARMACOLOGY Background Clinical studies have demonstrated that elevated levels of total cholesterol (total-C), low density lipoprotein cholesterol (LDL-C) and apolipoprotein B (Apo B), the major protein constituent of LDL, promote human atherosclerosis. In addition, decreased levels of high density lipoprotein cholesterol (HDL-C) are associated with the development of atherosclerosis. Epidemiologic studies have established that cardiovascular morbidity and mortality vary directly with the level of total-C and LDL-C and inversely with the level of HDL-C. Like LDL, cholesterol-enriched triglyceride-rich lipoproteins, including very-low- density lipoproteins (VLDL), intermediate-density lipoproteins (IDL), and remnants, can also promote atherosclerosis. The independent effect of raising HDL-C or lowering triglycerides (TG) on the risk of coronary and cardiovascular morbidity and mortality has not been determined.