A CB1 Receptor Antagonist As a Direct Interactional Partner for Μ- and Δ-Opioid Receptor
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Development of Swiss Biotechnology Beyond the Biopharmaceutical Sector in Memoriam Prof
BUILDING BRIDGES BETWEEN BIOTECHNOLOGY AND CHEMISTRY – IN MEMORIAM ORESTE GHISALBA CHIMIA 2020, 74, No. 5 345 doi:10.2533/chimia.2020.345 Chimia 74 (2020) 345–359 © H.P. Meyer and O. Werbitzky Development of Swiss Biotechnology Beyond the Biopharmaceutical Sector In memoriam Prof. Dr. Oreste Ghisalba (1946–2018) Hans-Peter Meyera* and Oleg Werbitzkyb Abstract: Although diverse, the potential business opportunities for biotechnology outside the biopharmaceutical market are very large. White biotechnology can offer sustainable operations and products, while investments tend to be lower than those in red biotechnology. But a number of bottlenecks and roadblocks in Switzerland must be removed to realise the full potential of white biotechnology. This was also the point of view of Oreste Ghisalba, who wanted to be part of a new initiative to facilitate the creation of additional business, new pro- cesses and new products. This initiative requires the identification and the use of synergies and a much better cooperation between academia and industry through targeted networking. Unfortunately, we must carry on with this task without Oreste, whom we will miss for his deep knowledge and friendship. Keywords: Bio-based · Fine chemicals · Industrial biotechnology · Swiss economy · White biotechnology Hans-Peter Meyer holds a PhD in micro- venture partners and Managing Director of NC Health Sciences biology from the University of Fribourg since 2015. (Switzerland). He spent three years post- graduate and postdoc studies at the STFI 1. A Short History of the Swiss Biotechnology Industry in Stockholm (Sweden), the University The Swiss pharmaceutical and chemical industries have been of Pennsylvania and Lehigh University in responsible for almost half of the country’s exports for many years the USA. -
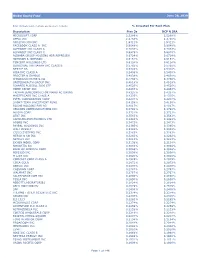
Global Equity Fund Description Plan 3S DCP & JRA MICROSOFT CORP
Global Equity Fund June 30, 2020 Note: Numbers may not always add up due to rounding. % Invested For Each Plan Description Plan 3s DCP & JRA MICROSOFT CORP 2.5289% 2.5289% APPLE INC 2.4756% 2.4756% AMAZON COM INC 1.9411% 1.9411% FACEBOOK CLASS A INC 0.9048% 0.9048% ALPHABET INC CLASS A 0.7033% 0.7033% ALPHABET INC CLASS C 0.6978% 0.6978% ALIBABA GROUP HOLDING ADR REPRESEN 0.6724% 0.6724% JOHNSON & JOHNSON 0.6151% 0.6151% TENCENT HOLDINGS LTD 0.6124% 0.6124% BERKSHIRE HATHAWAY INC CLASS B 0.5765% 0.5765% NESTLE SA 0.5428% 0.5428% VISA INC CLASS A 0.5408% 0.5408% PROCTER & GAMBLE 0.4838% 0.4838% JPMORGAN CHASE & CO 0.4730% 0.4730% UNITEDHEALTH GROUP INC 0.4619% 0.4619% ISHARES RUSSELL 3000 ETF 0.4525% 0.4525% HOME DEPOT INC 0.4463% 0.4463% TAIWAN SEMICONDUCTOR MANUFACTURING 0.4337% 0.4337% MASTERCARD INC CLASS A 0.4325% 0.4325% INTEL CORPORATION CORP 0.4207% 0.4207% SHORT-TERM INVESTMENT FUND 0.4158% 0.4158% ROCHE HOLDING PAR AG 0.4017% 0.4017% VERIZON COMMUNICATIONS INC 0.3792% 0.3792% NVIDIA CORP 0.3721% 0.3721% AT&T INC 0.3583% 0.3583% SAMSUNG ELECTRONICS LTD 0.3483% 0.3483% ADOBE INC 0.3473% 0.3473% PAYPAL HOLDINGS INC 0.3395% 0.3395% WALT DISNEY 0.3342% 0.3342% CISCO SYSTEMS INC 0.3283% 0.3283% MERCK & CO INC 0.3242% 0.3242% NETFLIX INC 0.3213% 0.3213% EXXON MOBIL CORP 0.3138% 0.3138% NOVARTIS AG 0.3084% 0.3084% BANK OF AMERICA CORP 0.3046% 0.3046% PEPSICO INC 0.3036% 0.3036% PFIZER INC 0.3020% 0.3020% COMCAST CORP CLASS A 0.2929% 0.2929% COCA-COLA 0.2872% 0.2872% ABBVIE INC 0.2870% 0.2870% CHEVRON CORP 0.2767% 0.2767% WALMART INC 0.2767% -

Selection and Characterization of Amyloid-Β1-42 Binding D-Enantiomeric Peptides for Potential Therapeutic Intervention of Alzheimer´S Disease
Selection and characterization of Amyloid-β1-42 binding D-enantiomeric peptides for potential therapeutic intervention of Alzheimer´s disease Inaugural-Dissertation zur Erlangung des Doktorgrades der Mathematisch-Naturwissenschaftlichen Fakultät der Heinrich-Heine-Universität Düsseldorf vorgelegt von Stephan Rudolph aus Dessau Düsseldorf, April 2015 Die vorliegende Arbeit wurde in der Zeit von April 2011 bis Februar 2015 am Institut für Physikalische Biologie der Heinrich-Heine-Universität Düsseldorf unter der Leitung von Prof. Dr. Dieter Willbold angefertigt. Gedruckt mit der Genehmigung der Mathematisch-Naturwissenschaftlichen Fakultät der Heinrich-Heine-Universität Düsseldorf Referent: Prof. Dr. Dieter Willbold Koreferent: Prof. Dr. Georg Groth Tag der mündlichen Prüfung: „Die Dummheiten wechseln, und die Dummheit bleibt.“ (Erich Kästner) „Die dunkelste Stunde ist die vor Sonnenaufgang.“ (chinesisches Sprichwort) Für Tante Elli Index Index Index I - IV 1. Introduction 1 1.1 Alzheimer´s disease (AD) – an overview .................................................... 1 1.1.1 General information ............................................................................. 1 1.1.2 History and pathology of AD ................................................................ 3 1.1.3 The amyloid precursor protein (APP) ................................................... 4 1.1.4 From APP to Aβ ................................................................................... 7 1.1.5 The physiological role of Aβ and Tau ................................................. -

Q4 2018 CDMO – Transaction Comps
Q4 2018 CDMO – Transaction Comps CDMO Transaction Comps USD in millions Announced Geographic Enterprise EV / LTM EV / LTM Date Target Target Description Buyer Location Value LTM Revenue LTM EBITDA Revenue EBITDA CDMO offering services including analytical testing, API and Nov-18 Avista Pharma Solutions1 Drug Product development, early stage discovery, and Cambrex Corporation USA $252.0 $65.0 NA 3.9x NA microbiology testing support Operates as a custom manufacturer of APIs and registered Jul-18 AMPAC Fine Chemicals LLC2 SK Holdings Co., Ltd. USA 455.0 200.0 NA NA NA intermediates for the pharmaceutical industry globally Provides contract development and manufacturing services in Jul-18 Halo Pharma, Inc. Cambrex Corporation USA 425.0 105.0 $27.0 4.0x 15.7x various dosage forms and drug delivery platforms Offers services in the areas of early-phase formulation Madison Dearborn Jun-18 Alcami Corporation USA NA NA NA NA NA development, analytical testing, and manufacturing Partners Provides contract development and manufacturing services to Catalent Pharma Sep-17 Cook Pharmica LLC USA 950.0 177.8 NA 5.3x NA pharmaceutical and biopharmaceutical companies Solutions, Inc. Provides drug discovery and development services as well as Jul-17 Aptuit LLC Evotec AG USA 300.0 92.9 11.6 3.2x 25.8x API development and manufacturing services Provides chemical and analytical development and custom Jun-17 PCAS SA Novacap France 335.9 243.7 19.7 1.4x 17.0x manufacturing services Contract research and manufacturing company that provides Jun-17 Albany Molecular Research, Inc. The Carlyle Group L.P. -

Inflammation As a Therapeutic Target for Alzheimer's
From DEPARTMENT OF NEUROBIOLOGY, CARE SCIENCE AND SOCIETY Karolinska Institutet, Stockholm, Sweden INFLAMMATION AS A THERAPEUTIC TARGET FOR ALZHEIMER’S DISEASE ERIK HJORTH Stockholm 2010 All previously published papers were reproduced with permission from the publisher. Published by Karolinska Institutet. Printed by [name of printer] The author may be contacted on [email protected] © Erik Hjorth, 2010 ISBN 978-91-7409-883-9 ABSTRACT Alzheimer‟s disease (AD) is a progressive neurodegenerative disorder which is characterised by impairment of memory and learning. The impairment is caused by neuronal death which originates in the parts of the brain that execute memory functions: the entorhinal cortex and hippocampus. The neuronal death is believed to be caused by the amyloid- (A ) peptide which is prone to oligomerisation and aggregation into insoluble amyloid plaques (AP). The levels of soluble A and the number of AP:s are increased in the AD brain which is attributed to increased production and impaired clearance of A . Another hallmark of AD, after neuronal death and the increased presence of A , is inflammation in the form of activated microglia and increased levels of inflammatory proteins in the brain. Inflammation in the CNS has been shown to increase the production of A and to impair, and even kill, neurons. On the other hand, inflammation has been shown to increase the removal of pathogens, such as A , from the brain by increasing the phagocytic acticity of microglia. Inflammation is also associated with an increased secretion of neurotrophic factors that can protect neurons. Somehow this clearance of A is impaired in AD and the levels of neurotrophic factors are decreased. -

Angiotensin-I-Converting Enzyme and Prolyl Endopeptidase Inhibitory Peptides from Marine Processing By-Products
instlttiild lctt«rkenny Talcneolaiochta institute Lyit Lelttr Ccanalnn of Technology Angiotensin-I-converting enzyme and prolyl endopeptidase inhibitory peptides from marine processing by-products Julia Wilson Supervisor: Dr. B. Camey, Letterkenny Institute of Technology External Supervisor: Dr, M. Hayes, Teagasc. Ash town, Dublin Submitted to the Higher Education and Training Awards Council in fulfilment of the requirements for the degree of Master of Science by research. Table of Contents Declaration 3 Abstract 4 List of Abbreviations 6 List of Figures 8 List of Tables 10 Publications 11 Acknowledgements 12 Chapter 1: Literature review 1.1 Introduction A 13 1.2 Mackerel and Whelk life history and habitats 14 1.3 Function of ACE-I and PEP inhibitory peptides 16 1.4 Sources of ACE-I and PEP inhibitory peptides 20 1.5 Derivatisation of ACE-I and PEP inhibitory peptides 22 1.5.1 Principles of capillary electrophoresis (CE) 28 1.5.2 Principles of high performance liquid chromatography (HPLC) 31 1.5.3 Principles of mass spectrometry (MS) 33 1.6 Structural properties involved in ACE-I and PEP inhibitory 33 activities of peptides 1.7 Bioactive peptides as functional foods 35 1.7.1 Survival of bioactive peptide inhibitors in vivo 36 1.8 Aims and objectives 38 Chapter 2: Materials and Methods 2.1 General materials and methods 40 2.1.1 Chemicals and reagents 40 2.1.2 Buffer preparation 41 2.1.3 Bradford protein assay 42 1 2.2 Enzyme hydrolytic studies 43 2.2.1 Sample pre-treatments 43 2.2.2 Hydrolytic enzymes and hydrolytic reactions 45 2.3 Colorimetric -

Investigation of the Novel Hormone Kisspeptin in Disorders Of
Investigation of the novel hormone kisspeptin in disorders of reproduction Gurjinder Monica Kaur Nijher Department of Investigative Medicine Thesis submitted for the degree of Doctor of Philosophy Imperial College London 2012 1 Abstract The novel hormone kisspeptin has been identified to play a pivotal role in the regulation of the hypothalamo-pituitary-gonadal axis. Previous studies from our laboratory have demonstrated that a bolus administration of kisspeptin-54 can acutely stimulate gonadotrophin release in healthy men and women. However, no previous studies have examined the effects of kisspeptin-54 to women with infertility. In this study I have examined the effects of acute and chronic administration of kisspeptin-54 on women with infertility due to hypothalamic amenorrhoea. This study has identified that acute administration of kisspeptin to women with hypothalamic amenorrhoea results in stimulation of reproductive hormones, but chronic administration for two weeks of twice daily injections of kisspeptin-54 at a dose of 6.4 nmol/kg, results in tachyphylaxis. I have conducted further studies to examine the time course of desensitisation of the kisspeptin receptor. I have also determined that a dosing regime of twice weekly administration of kisspeptin-54 results in sustained stimulation of gonadotrophin release. I have performed the first study of kisspeptin-10 administration to men and women. These results demonstrate that kisspeptin-10 stimulates gonadotrophin release in men as well as women during the preovulatory phase of the menstrual cycle; but fails to stimulate gonadotrophin release in women during the follicular phase. This reveals a sexual dimorphism in response to the kisspeptin-10. These findings have important ramifications for the potential use of kisspeptin-54 and kisspeptin-10 as a therapeutic agent in disorders of reproduction. -

The Role of Kisspeptin in Reproduction
The Role of Kisspeptin in Reproduction Thesis submitted for the degree of Doctor of Philosophy Imperial College London Alexander Comninos 2014 Section of Investigative Medicine Division of Diabetes, Endocrinology & Metabolism Imperial College London 1 Declaration of Copyright ___________________________________________ The copyright of this thesis rests with the author and is made available under a Creative Commons Attribution Non-Commercial No Derivatives Licence. Researchers are free to copy, distribute or transmit the thesis on the condition that they attribute it, that they do not use it for commercial purposes and that they do not alter, transform or build upon it. For any reuse or redistribution, researchers must make clear to others the licence terms of this work. 2 Abstract ___________________________________________ Kisspeptin is a recently identified hypothalamic neuropeptide hormone which is a critical regulator of mammalian reproductive physiology. In addition, kisspeptin administration stimulates gonadotrophin release in all species tested. Kisspeptin may therefore serve as a potential therapy for reproductive pathologies. The role of kisspeptin has been studied extensively within the hypothalamus but little is known about its extra-hypothalamic effects. Using manganese-enhanced MRI to detect neuronal activity, I demonstrate marked decreases in neuronal activity in the amygdala of adult mice after kisspeptin administration. This data reveals a novel effect of kisspeptin with pharmacological implications. The human kisspeptin peptides kisspeptin-10, -13, -14, and -54 are named according to their constituent amino acids. Kisspeptin-10 is the smallest and most easily synthesised peptide from a pharmacological point of view. By investigating the effects of the kisspeptin-10 peptide in healthy men and women, I uncover a sexual dimorphism in gonadotrophin response to kisspeptin-10 as well as a greater potency of kisspeptin-54 over kisspeptin-10 in healthy women. -

Amyloid Β Protein Negatively Regulates the Human Platelet Activation Induced by Thrombin Receptor- Activating Protein
Amyloid β protein negatively regulates the human platelet activation induced by thrombin receptor- activating protein Daisuke Mizutani Department of Neurosurgery Gifu University Graduate School of Medicine Haruhiko Tokuda Department of Clinical Labolatory/Medical Genome Center Biobank, National Center for Geriatrics and gerontology Takashi Onuma Department of Anethesiology and Pain Medicine, Gifu University Graduate School of Medicine Kodai Uematsu Department of Neurosurgery, Gifu University Graduate School of Medicine Daiki Nakashima Department of Anesthesiology and Pain Medicine, Gifu University Graduate School of Medicine Kyohei Ueda Department of Anesthesiology and Pain Medicine, Gifu University Graduate School of Medicine Tomoaki Doi Department of Emergency and Disaster Medicine, Gifu University Graduate School of Medicine Yukiko Enomoto Department of Neurosurgery, Gifu University Graduate School of Medicine Rie Matsushima-Nishiwaki Department of Pharmacology, Gifu University Graduate School of Medicine Shinji Ogura Department of Emergency and Disaster Medicine, Gifu University Graduate School of Medicine Hiroki Iida Department of Anesthesia and Pain Medicine, Gifu University Graduate School of Medicine Osamu Kozawa ( [email protected] ) Department of Pharmacology, Gifu University Graduate School of Medicine https://orcid.org/0000- 0002-4834-6073 Toru Iwama Department of Neurosurgery, Gifu University Graduate School of Medicine Research Page 1/22 Keywords: Amyloid β protein, platelet, TRAP, PDGF, HSP27, cerebral amyloid angiopathy Posted Date: April 9th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-391777/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 2/22 Abstract Background: Amyloid β protein (Aβ) is the main product derived from amyloid precursor protein (APP) by sequential enzymatic actions. -

Peripher Injiziertes Sulfatiertes Cholezystokinin-Oktapeptid Aktiviert
Aus der Klinik mit Schwerpunkt Psychosomatik der Medizinischen Fakultät Charité - Universitätsmedizin Berlin DISSERTATION Peripher injiziertes sulfatiertes Cholezystokinin-Oktapeptid aktiviert Cocaine- and amphetamine-regulated transcript- immunreaktive Neurone des Nucleus paraventricularis in Ratten zur Erlangung des akademischen Grades Doctor medicinae (Dr. med.) vorgelegt der Medizinischen Fakultät Charité – Universitätsmedizin Berlin von Lisa Peter aus Adenau Datum der Promotion: 5. Dezember 2014 Inhaltsverzeichnis Inhaltsverzeichnis Abstract Abkürzungsverzeichnis 1. Einleitung 1 1.1 Die Regulation der Nahrungsaufnahme 1 1.2 Die zentrale Nahrungsregulation im Hypothalamus und 2 Hirnstamm 1.2.1 Der Nucleus arcuatus 5 1.2.2 Der Nucleus paraventricularis 6 1.2.3 Der Nucleus tractus solitarius 7 1.3 Die nahrungsregulierenden Botenstoffe 8 1.3.1 Die peripheren nahrungsregulierenden Botenstoffe 9 1.3.1.1 Das Peptidhormon Cholezystokinin 10 1.3.1.1.1 Die Entdeckung von Cholezystokinin 10 1.3.1.1.2 Die molekulare Struktur des Cholezystokinin 10 1.3.1.1.3 Die Wirkung von Cholezystokinin auf die Nahrungsaufnahme 11 1.3.1.1.4 Die Cholezystokinin-Rezeptoren 12 1.3.1.1.5 Die nervale Informationsvermittlung von Cholezystokinin 12 1.3.1.1.6 Das zentrale Nervensystem als Wirkungsort des Cholezystokinin 13 1.3.1.1.7 Die weiteren Funktionen des Cholezystokinin 14 1.3.2 Die zentralen nahrungsregulierende Botenstoffe 14 1.3.2.1 Der Neuromodulator Cocaine- and amphetamine-regulated 16 transcript 1.3.2.1.1 Die Lokalisation des CART-Peptids 16 1.3.2.1.2 Die Wirkung des CART-Peptids auf die Nahrungsaufnahme 18 1.3.2.1.3 Die weiteren Funktionen des CART-Peptids und der CART- 18 mRNA 1.3.2.2 Das Peptidhormon Corticotropin-releasing factor 19 1.3.2.3 Das Peptidhormon Oxytocin 20 1.4 Das Protoonkogen c-Fos als Marker neuronaler Aktivität 21 1.5 Die Zielsetzung 22 Inhaltsverzeichnis 2. -

Peptider Som Läkemedel En Marknads- Och Trendanalys
17-X5 Peptider som läkemedel En marknads- och trendanalys Elina Annala, Matilda Brink, Simon Ekdahl, Sofia Iggström, Felicia Mejàre, Ebba Spetsare Beställare: Bio-Works Technologies AB Beställarrepresentant: Johan Ledin Handledare: Lena Henriksson 1MB332, Självständigt arbete i molekylär bioteknik, 15 hp, vt 2017 Civilingenjörsprogrammet i molekylär bioteknik Institutionen för biologisk grundutbildning, Uppsala universitet Sammanfattning Rapporten syftar till att ge en ¨oversk˚adligbild av den aktuella peptidbaserade l¨akemedelsmarknaden. Fr˚anen i rapporten definierad marknad har prim¨ardata kring etablerade f¨oretag p˚a marknaden, samt peptidbaserade l¨akemedel till f¨ors¨aljning p˚amarknaden, sammanst¨allts. Rapporten finner att onkologiska- och metaboliska sjukdomar ¨ar ¨overrepresenterade i statistiken, vilket ¨aven st¨ods av rapporter fr˚antredje part. Utforskandet av samarbeten mellan f¨oretag och akademi, f¨oretag och f¨oretag, och andra samarbetsformer presenteras ocks˚a.En tydlig trend i dessa unders¨okningar tyder p˚aatt kontraktsbaserade samarbeten mellan organisationer, d¨ar forskning och produktion g¨ors av ett externt f¨oretag som ett annat f¨oretag anv¨ander sig av, ¨ar p˚a uppg˚ang. Detta s¨anker kostnader f¨or framst¨allningen av nya l¨akemedel, och effektiviserar denna process. Trender inom forskningen talar ocks˚a f¨or att folkh¨alsosjukdomar i v¨astv¨arlden ¨ar trends¨attande f¨or l¨akemedelsf¨oretagens inriktningar. Rapporten baseras p˚aprim¨art insamlad statistik som verifieras genom rapporter fr˚antredje part, fallstudier f¨or att exemplifiera generaliseringar gjorda av andra k¨allor, samt reflektioner kring vad denna information utr¨onar i f¨or slutsats. Inneh˚all Ansvarstagande . .1 Projektbest¨allning . .2 Bio-Works Technologies . -
WO 2011/053831 Al
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date 5 May 2011 (05.05.2011) WO 2011/053831 Al (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every CI2Q 1/68 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, (21) Number: International Application CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, PCT/US20 10/0548 10 DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, 29 October 2010 (29.10.2010) KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, (25) Filing Language: English NO, NZ, OM, PE, PG, PH, PL, PT, RO, RS, RU, SC, SD, (26) Publication Language: English SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: 61/256,7 17 30 October 2009 (30.10.2009) US (84) Designated States (unless otherwise indicated, for every 61/264,588 25 November 2009 (25.1 1.2009) US kind of regional protection available): ARIPO (BW, GH, 61/326,1 76 20 April 2010 (20.04.2010) US GM, KE, LR, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, (71) Applicant (for all designated States except US): TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, PROMETHEUS LABORATORIES INC.