Novel Fluorous Hybrid Surface Modification Characterized by Wetting Dynamics, Morphology and Nanomechanics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Inverse Gas Chromatographic Examination of Polymer Composites
Open Chem., 2015; 13: 893–900 Invited Paper Open Access Adam Voelkel*, Beata Strzemiecka, Kasylda Milczewska, Zuzanna Okulus Inverse Gas Chromatographic Examination of Polymer Composites DOI: 10.1515/chem-2015-0104 received December 30, 2014; accepted April 1, 2015. of composite components and/or interactions between them, and behavior during technological processes. This paper reviews is the examination of Abstract: Inverse gas chromatographic characterization various polymer-containing systems by inverse gas of resins and resin based abrasive materials, polymer- chromatography. polymer and polymer-filler systems, as well as dental restoratives is reviewed. Keywords: surface activity, polymer-polymer interactions, 2 Discussion adhesion, dental restoratives, inverse gas chromatography 2.1 Surface energy and adhesion 1 Introduction IGC is useful for surface energy determination of solid polymers and fillers. Solid surface energy of consists of Inverse gas chromatography (IGC) was introduced in 1967 dispersive ( , from van der Waals forces) and specific by Kiselev [1], developed by Smidsrød and Guillet [2], and ( , from acid-base interactions) components: is still being improved. Its popularity is due to its simplicity D and user friendliness. Only a standard gas chromatograph = + (1) is necessary [3] although more sophisticated equipment has been advised. “Inverse” relates to the aim of the experiment. for solids can be calculated according to several It is not separation as in classical GC, but examination of methods [7]; one of the most often used is that of Schultz- the stationary phase properties. Test compounds with Lavielle [8-12]: known properties are injected onto the column containing dd the material to be examined. Retention times and peak RT××ln VN =× 2 N ×× aggsl × + C (2) profiles determine parameters describing the column filling. -

Inverse Gas Chromatography in the Examination of Adhesion Between
www.nature.com/scientificreports OPEN Inverse gas chromatography in the examination of adhesion between tooth hard tissues and restorative dental materials Zuzanna Buchwald1*, Beata Czarnecka2 & Adam Voelkel1 The adhesion is a crucial issue in the bonding of dental restorative materials to tooth hard tissues. A strong and durable bond between artifcial and natural materials is responsible for the success of the restoration in the oral cavity; therefore it has to be thoroughly examined before new restorative material is introduced to the market and used clinically. Among all methods used to examine bonding strength, most of them require a large number of healthy teeth to be conducted. In this paper, the bond strength between tooth hard tissues (dentin and enamel) and an exemplary restorative composite was examined with the non-conventional method, i.e. inverse gas chromatography. Dentin and enamel from bovine teeth were separated and subjected to the standard preparation procedure using the 3-component etch-and-rinse commercial bonding system. Tissues, as well as commercial restorative composite, were examined using inverse gas chromatography. The work of adhesion between dentin/enamel and composite was calculated. Obtained results were compared with the values of shear bond strength of six confgurations, i.e. etched dentin/enamel-composite, primed dentin/enamel-composite, and bonded dentin/enamel-composite. All obtained results proved that there is a correlation between the values describing bond strength obtained from inverse gas chromatography and direct mechanical tests (shear bond strength tests). It proves that inverse gas chromatography is a powerful perspective tool for the examination of bond strength between tooth hard tissues and potential dental materials without using a large number of health tooth tissues. -

Wettability of Mineral and Metallic Powders: Applicability and Limitations of Sessile Drop Method and Washburn's Technique (2012) Powder Technology, 226, 6877
Susana, L., Campaci, F., Santomaso, A.C. Wettability of mineral and metallic powders: Applicability and limitations of sessile drop method and Washburn©s technique (2012) Powder Technology, 226, 68-77. (DOI: 10.1016/j.powtec.2012.04.016) Wettability of mineral and metallic powders: applicability and limitations of sessile drop method and Washburn’s technique Laura Susanaa, Filippo Campacib, Andrea C. Santomasoa,* a DII – Department of Industrial Engineering, University of Padova Via Marzolo 9, 35131, Padova, Italy b Fileur® - Trafilerie di Cittadella, Via Mazzini 69, 35013, Cittadella, Italy Corresponding author: Andrea Santomaso; email: [email protected] Keywords: Wettability; Contact angle; Minerals powders; Washburn's method Abstract Characterization of powder wettability is a prerequisite to the understanding of many processes of industrial relevance such as agglomeration which spans from pharmaceutical and food applications to metallurgical ones. The choice of the wetting fluid is crucial: liquid must wet the powder in order for agglomeration to be successful. Different methods for wettability assessment of powders were reported in the literature, however the sessile drop method and capillary rise test remain among the most widely employed because they are easy to perform and inexpensive. In this paper, the application and limitations of sessile drop method and capillary rise test on mineral and metallic surface were discussed. This work provides a collection of wettability measurements using several powders and liquid binders which are involved in the manufacturing process of welding wires. Moreover a new reference liquid for the calibration of capillary rise method was proposed. 1. Introduction Wet granulation of fine powders is carried out in many industries, from pharmaceutical and agrochemical industries to mineral and metallurgical ones, to improve physical and material properties of powders such as flowability, rate of dissolution and robustness to segregation [1–3]. -

Experimental and Computational Methods Pertaining to Surface Tension of Pharmaceuticals
3 Experimental and Computational Methods Pertaining to Surface Tension of Pharmaceuticals Abolghasem Jouyban1 and Anahita Fathi-Azarbayjani2 1Drug Applied Research Center and Faculty of Pharmacy, Tabriz University of Medical Sciences, Tabriz, 2Faculty of Medicine, Urmia University of Medical Sciences, Urmia, Iran 1. Introduction The molecules of a fluid experiences attractive forces exerted on it by all its neighboring molecules. In the bulk of the liquid, molecules are attracted equally in all directions resulting in a net force of zero. Molecules at or near the surface experience attractive force which tends to pull them to the interior. Surface chemistry deals with thermodynamic and kinetic parameters that take place between two different coexisting phases at equilibrium. Surface tension, γ is free energy of the surface at any air/fluid interface defined as force per unit length or energy per unit area. The latter term, also called surface energy, is more useful in thermodynamics and it applies to solids as well as liquid surfaces. The surface free energy of a liquid is measured by its surface tension and the surface free energy of a solid can be revealed by contact angle measurements. The surface tension measurement depends very markedly upon the presence of impurities in the liquid, temperature and pressure changes (Buckton, 1988). Surface tension is a phenomenon that we see in our everyday life. Human biological fluids, e.g. serum, urine, gastric juice, amniotic liquid, cerebrospinal and alveolar lining liquid contain numerous low-and high-molecular weight surfactants, proteins and lipids that adsorb at liquid interface. The physicochemical processes that take place in these interfaces are extremely important for the vital function of body organs and have a great impact on pharmacodynamic parameters of drug molecules (Kazakov et al., 2000; Trukhin et al., 2001). -

The University of Hull Interaction of Oils with Surfactants Being a Thesis
The University of Hull Interaction of oils with Surfactants being a thesis submitted for the Degree of Doctor of Philosophy in the University of Hull by Jamie Robert MacNab B. Sc. (Hons) November 1996 Acknowledgements I would like to thank my three supervisors, Dr Bernie Binks, Professor Paul Fletcher and Professor Robert Aveyard, for their guidance, support and advice during my PhD. Also many thanks to Dr John Clint of the Surfactant Science Group at the University of Hull for contributing his valued advice. I am grateful to Unilever Research, Port Sunlight Laboratories, for funding the project, and many thanks also to Drs Abid Khan-Lodhl and Tim Finch for participating in several fruitful discussions during the project. I also appreciate the help given by Chris Maddison and Marie Betney at Unilever Research for their advice and guidance during my time at Port Sunlight and also thanks to the technical staff of Hull University Chemistry Department. Finally, many thanks to the other members of the Surfactant Science Research Group for making my time in the group so enjoyable. Abstract This thesis is concerned with the interactions of oils with surfactants. The understanding of these interactions is important for a number of practical applications which include perfume delivery in fabric softeners. Both non-ionic surfactants, of the general formula H( CH2)n( OCH2CH2)m-OH, and lome surfactants, of the general formula CnH2n+lW(CH3)3Br" were used in the study. Oils of varying polarity were investigated from non-polar alkane oils to moderately polar perfume oils. Initially, the work of adhesions of the perfume oils with water were studied to establish where these oils 'fitted-in' to a range of oils of varying polarity. -

Surface Treatment of High Density Polyethylene (HDPE) Film Using Atmospheric Pressure Dielectric Barrier Discharge (APDBD) in Air and Argon
International Research Journal of Engineering Science, Technology and Innovation (IRJESTI) Vol. 2(1) pp. 1-7, January, 2013 Available online http://www.interesjournals.org/IRJESTI Copyright © 2013 International Research Journals Full Length Research Paper Surface treatment of high density polyethylene (HDPE) film using atmospheric pressure dielectric barrier discharge (APDBD) in air and argon *Joshi Ujjwal Man and Subedi Deepak Prasad Department of Natural Sciences, School of Science, Kathmandu University P. O. Box No. 6250, Dhulikhel, Kathmandu, Nepal Accepted January 18, 2013 Thin films of high density polyethylene (HDPE) were treated for improving hydrophilicity using non- thermal plasma generated by line frequency atmospheric pressure dielectric barrier discharge (APDBD) in air and argon. HDPE samples before and after the treatments were studied using contact angle measurements and scanning electron microscopy (SEM). Distilled water (H 2O), glycerol (C 3H8O3) and diiodomethane (CH 2I2) were used as test liquids. The contact angle measurements between test liquids and HDPE samples were used to determine total surface free energy using sessile drop technique. Ageing effect was characterized by means of contact angle measurements versus ageing time. It was found that the treatment of HDPE in argon seems to be more stable and uniform with higher roughness with respect to APDBD treatment time than the treatment of HDPE in air. Keywords: Thin films, polymer, atmospheric pressure dielectric barrier discharge, hydrophilicity, sessile drop technique. INTRODUCTION Polymers are very often used as films and foils for processes to large-scale objects, especially under the packaging, protective coatings, sealing applications, etc. condition of continuous treatment. Recently, much A low surface energy may be desirable in them for attention has been paid to the use of non-thermal several applications, but for other applications it is a plasmas under atmospheric pressure for surface disadvantage, which has to overcome Sira et al., (2005). -

Is It Important to Know the Surface Free Energy of Textiles? Methods to Characterize Textile/Fibre Surface for Different Treatments Like Printing, Dyeing, Coating Etc
Is it important to know the surface free energy of textiles? Methods to characterize textile/fibre surface for different treatments like printing, dyeing, coating etc. R. Minch Krüss GmbH, Borsteler Chaussee 85, 22453 Hamburg, Germany. Email: [email protected] Wettability and adhesion as well as water- or dirt-repellent properties are determined by surface energy. This plays a key role in problems concerning hydrophilicity/hydrophobicity and optimization of printing, coating, dyeing and cleaning of textiles. This presentation describes existing practical ways of surface free energy determination on different textile surfaces like fibres, fibre bundles, woven/nonwoven textiles and fabrics. There are two different measurement techniques which can be applied for this purpose: optical and non optical. The optical method is based on standard sessile drop technique and can be used mostly for fabrics. The study of wetting behaviour of textiles using this method can hardly be influenced due to unevenness of the sample and rapid ad- or absorption of test liquids. This can be remedied by the captive bubble method. Non optical methods are based on using balance based tensiometers. Here two different methods can be distinguished as well: dynamic Wilhelmy method which is usually applied for single fibres and Washburn method which can be applied for characterization of fibre bundles and woven textiles. Advantages and limitations of these methods will be discussed. 1. Why we need to know surface free energy of fibres and/or textiles? Fibres affect all of us in one way or another during our daily lives. They may be used in the form of a composite material, a woven or non-woven technical textile or even in reinforced concrete structures. -

Inverse Gas Chromatography with Film Cell Unit: an Attractive Alternative Method to Characterize Surface Properties of Thin Films Géraldine L
Inverse Gas Chromatography with Film Cell Unit: An Attractive Alternative Method to Characterize Surface Properties of Thin Films Géraldine L. Klein, Pierre G, Marie-Noëlle Bellon-Fontaine, Marianne Graber To cite this version: Géraldine L. Klein, Pierre G, Marie-Noëlle Bellon-Fontaine, Marianne Graber. Inverse Gas Chro- matography with Film Cell Unit: An Attractive Alternative Method to Characterize Surface Proper- ties of Thin Films. Journal of Chromatographic Science, Oxford University Press (OUP): Policy F, 2015, 53 (8), pp.1233-1238. 10.1093/chromsci/bmv008. hal-01247054 HAL Id: hal-01247054 https://hal.archives-ouvertes.fr/hal-01247054 Submitted on 21 Dec 2015 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. 1 1 Inverse Gas Chromatography with film cell unit: an attractive alternative method to 2 characterize surface properties of thin films. 3 4 Klein, G.L. 1, Pierre, G. 1, Bellon-Fontaine, M.N. 2, Graber, M. 1* 5 6 (1) UMR 7266 CNRS - ULR LIENSs, Equipe Approches Moléculaires Environnement Santé. Université de La 7 Rochelle, UFR Sciences, Bâtiment Marie Curie, avenue Michel Crépeau, 17042 La Rochelle, France. 8 (2) UMR 0763 – MICALIS - Agro-ParisTech-INRA – Equipe Bioadhésion-Biofilm et Hygiène des Matériaux, 25 9 avenue de la république, 91300 Massy, France. -

In Vitro Analysis of Wettability and Physical Properties of Blister Pack
In vitro analysis of wettability and physical properties of blister pack solutions of hydrogel contact lenses by Kara Laura Menzies A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree of Master of Science in Vision Science Waterloo, Ontario, Canada, 2010 © Kara Laura Menzies 2010 AUTHOR’S DECLARATION I hereby declare that I am the sole author of this thesis. This is a true copy of the thesis, including any required final revisions, as accepted by my examiners. I understand that my thesis may be made electronically available to the public. ii Abstract Contact lens success is primarily driven by comfort of the lens in eye. Over the years, many modifications have been made to the lens surface and bulk material to improve comfort of the lens, however 50% of contact lens wearers still report dry eye symptoms while wearing their lenses. Wettability of the lens material plays a large role in lens comfort, primarily due to its influence in tear film stability. In vitro wettability of contact lenses has typically been assessed by measuring the water contact angle on the lens surface. Currently there are three techniques to measure the in vitro wettability of contact lenses, the sessile drop technique, captive bubble technique, and the Wilhelmy balance method. To date, there is much published on assessing wettability using the sessile drop and captive bubble technique, however there is no data published looking at the in vitro wettability of hydrogel contact lenses measured by the Wilhelmy balance method. Accumulation and deposition of tear components on the lens surface can also affect lens performance, by altering the wettability of the lens surface and causing lens spoilage. -

Dynamic Measurement of Low Contact Angles by Optical Microscopy
This is a repository copy of Dynamic Measurement of Low Contact Angles by Optical Microscopy. White Rose Research Online URL for this paper: http://eprints.whiterose.ac.uk/138705/ Version: Accepted Version Article: Campbell, JM and Christenson, HK orcid.org/0000-0002-7648-959X (2018) Dynamic Measurement of Low Contact Angles by Optical Microscopy. ACS Applied Materials and Interfaces, 10 (19). pp. 16893-16900. ISSN 1944-8244 https://doi.org/10.1021/acsami.8b03960 © 2018 American Chemical Society. This is an author produced version of a paper published in ACS Applied Materials and Interfaces. Uploaded in accordance with the publisher's self-archiving policy. Reuse Items deposited in White Rose Research Online are protected by copyright, with all rights reserved unless indicated otherwise. They may be downloaded and/or printed for private study, or other acts as permitted by national copyright laws. The publisher or other rights holders may allow further reproduction and re-use of the full text version. This is indicated by the licence information on the White Rose Research Online record for the item. Takedown If you consider content in White Rose Research Online to be in breach of UK law, please notify us by emailing [email protected] including the URL of the record and the reason for the withdrawal request. [email protected] https://eprints.whiterose.ac.uk/ Dynamic Measurement of Low Contact Angles by Optical Microscopy James M. Campbell*, Hugo K. Christenson School of Physics and Astronomy, University of Leeds, Woodhouse Lane, Leeds, LS2 9JT, United Kingdom. *Email: [email protected] Keywords: droplets; optics; wetting; wettability; mica. -
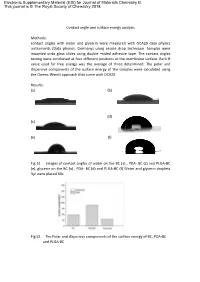
Contact Angles with Water and Glycerin Were Measured with OCA20 Data Physics Instruments (Data Physics, Germany) Using Sessile Drop Technique
Electronic Supplementary Material (ESI) for Journal of Materials Chemistry B. This journal is © The Royal Society of Chemistry 2018 Contact angle and surface energy analysis Methods: contact angles with water and glycerin were measured with OCA20 data physics instruments (Data physics, Germany) using sessile drop technique. Samples were mounted onto glass slides using double –sided adhesive tape. The contact angles testing were conducted at four different positions at the membrane surface. Each θ value used for free energy was the average of three determined. The polar and dispersive components of the surface energy of the samples were calculated using the Owens-Wendt approach that come with OCA20. Results: (a) (b) (d) (c) (e) (f) Fig S1. Images of contact angles of water on the BC (a) , PDA- BC (c) and PLGA-BC (e), glycerin on the BC (a) , PDA- BC (d) and PLGA-BC (f).Water and glycerin droplets 3μl were placed 60s. Fig S2. The Polar and dispersive components of the surface energy of BC, PDA-BC and PLGA-BC The contact angles of water and glycerin on various surface were measured respectively as shown in Fig.1. The dispersion and polar surface tension components of the liquids which were used to obtain the components of surface energy of the various samples are shown in Fig.2, suggesting that both dispersion and polar components increased after modification of PDA. As expected, the decrease in the surface energy, especially the polar component, can be observed in PLGA-BC membrane. Discussion BC membrane is composed of nano-size fibres and therefore possess a very high surface energy. -

Contact Angle Temperature Dependence for Water Droplets on Practical Aluminum Surfaces
Inf. J. Hem Moss Trmfir. Vol. 40, No. 5, pp. 1017-1033, 1997 Copyright 0 1996 Elswier Science Ltd Pergamon Printed in Great Britain. All rights reserved 0017-9310/97 s17.OO+o.oo PI1 : soo17-9310(%)00184-6 Contact angle temperature dependence for water droplets on practical aluminum surfaces JOHN ID. BERNARDIN, ISSAM MUDAWAR,_F CHRISTOPHER B. WALSHf and ELIAS I. FRANSESI Boiling ;and Two-phase Flow Laboratory, School of Mechanical Engineering, Purdue University, West Lafayette, IN 47907, U.S.A. (Received 2 April 1996 and injinalform 20 May 1996) Abstract-This paper presents an experimental investigation of the temperature dependence of the quasi- static advancing contact angle of water on an aluminum surface polished in accordance with surface preparation techniques commonly employed in boiling heat transfer studies. The surface, speculated to contain aluminum oxide and organic residue left behind from the polishing process, was characterized with scanning elelctron microscopy, surface contact profilometry, and ellipsometry. By utilizing a pressure vessel to raise the liquid saturation temperature, contact angles were measured with the sessile drop technique for surface temperatures ranging from 25 to 170°C and pressures from 101.3 to 827.4 kPa. Two distinct temperature-dependent regimes were observed. In the lower temperature regime, below 120°C a relatively constant contact angle of 90” was observed. In the high temperature regime, above 12O”C, the contact angle decreased in a fairly linear manner. Empirical correlations were developed to describe this behavior which ernula.ted previous experimental data for nonmetallic surfaces as well as theoretical trends. Copyright 0 1996 Elsevier Science Ltd.