Brimonidine Tartrate
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Brimonidine Tartrate; Brinzolamide
Contains Nonbinding Recommendations Draft Guidance on Brimonidine Tartrate ; Brinzolamide This draft guidance, when finalized, will represent the current thinking of the Food and Drug Administration (FDA, or the Agency) on this topic. It does not establish any rights for any person and is not binding on FDA or the public. You can use an alternative approach if it satisfies the requirements of the applicable statutes and regulations. To discuss an alternative approach, contact the Office of Generic Drugs. Active Ingredient: Brimonidine tartrate; Brinzolamide Dosage Form; Route: Suspension/drops; ophthalmic Strength: 0.2%; 1% Recommended Studies: One study Type of study: Bioequivalence (BE) study with clinical endpoint Design: Randomized (1:1), double-masked, parallel, two-arm, in vivo Strength: 0.2%; 1% Subjects: Males and females with chronic open angle glaucoma or ocular hypertension in both eyes. Additional comments: Specific recommendations are provided below. ______________________________________________________________________________ Analytes to measure (in appropriate biological fluid): Not applicable Bioequivalence based on (95% CI): Clinical endpoint Additional comments regarding the BE study with clinical endpoint: 1. The Office of Generic Drugs (OGD) recommends conducting a BE study with a clinical endpoint in the treatment of open angle glaucoma and ocular hypertension comparing the test product to the reference listed drug (RLD), each applied as one drop in both eyes three times daily at approximately 8:00 a.m., 4:00 p.m., and 10:00 p.m. for 42 days (6 weeks). 2. Inclusion criteria (the sponsor may add additional criteria): a. Male or nonpregnant females aged at least 18 years with chronic open angle glaucoma or ocular hypertension in both eyes b. -

Table 1. Glaucoma Medications: Mechanisms, Dosing and Precautions Brand Generic Mechanism of Action Dosage/Avg
OPTOMETRIC STUDY CENTER Table 1. Glaucoma Medications: Mechanisms, Dosing and Precautions Brand Generic Mechanism of Action Dosage/Avg. % Product Sizes Side Effects Warnings Reduction CHOLINERGIC AGENTS Direct Pilocarpine (generic) Pilocarpine 1%, 2%, 4% Increases trabecular outflow BID-QID/15-25% 15ml Headache, blurred vision, myopia, retinal detachment, bronchiole constriction, Angle closure, shortness of breath, retinal narrowing of angle detachment Indirect Phospholine Iodide (Pfizer) Echothiophate iodide 0.125% Increases trabecular outflow QD-BID/15-25% 5ml Same as above plus cataractogenic iris cysts in children, pupillary block, Same as above, plus avoid prior to any increased paralysis with succinylcholine general anesthetic procedure ALPHA-2 AGONISTS Alphagan P (Allergan) Brimonidine tartrate 0.1%, 0.15% with Purite Decreases aqueous production, increases BID-TID/up to 26% 5ml, 10ml, 15ml Dry mouth, hypotension, bradycardia, follicular conjunctivitis, ocular irritation, Monitor for shortness of breath, dizziness, preservative uveoscleral outflow pruritus, dermatitis, conjunctival blanching, eyelid retraction, mydriasis, drug ocular redness and itching, fatigue allergy Brimonidine tartrate Brimonidine tartrate 0.15%, 0.2% Same as above Same as above 5ml, 10ml Same as above Same as above (generic) Iopidine (Novartis) Apraclonidine 0.5% Decreases aqueous production BID-TID/up to 25% 5ml, 10ml Same as above but higher drug allergy (40%) Same as above BETA-BLOCKERS Non-selective Betagan (Allergan) Levobunolol 0.25%, 0.5% Decreases -

Title 16. Crimes and Offenses Chapter 13. Controlled Substances Article 1
TITLE 16. CRIMES AND OFFENSES CHAPTER 13. CONTROLLED SUBSTANCES ARTICLE 1. GENERAL PROVISIONS § 16-13-1. Drug related objects (a) As used in this Code section, the term: (1) "Controlled substance" shall have the same meaning as defined in Article 2 of this chapter, relating to controlled substances. For the purposes of this Code section, the term "controlled substance" shall include marijuana as defined by paragraph (16) of Code Section 16-13-21. (2) "Dangerous drug" shall have the same meaning as defined in Article 3 of this chapter, relating to dangerous drugs. (3) "Drug related object" means any machine, instrument, tool, equipment, contrivance, or device which an average person would reasonably conclude is intended to be used for one or more of the following purposes: (A) To introduce into the human body any dangerous drug or controlled substance under circumstances in violation of the laws of this state; (B) To enhance the effect on the human body of any dangerous drug or controlled substance under circumstances in violation of the laws of this state; (C) To conceal any quantity of any dangerous drug or controlled substance under circumstances in violation of the laws of this state; or (D) To test the strength, effectiveness, or purity of any dangerous drug or controlled substance under circumstances in violation of the laws of this state. (4) "Knowingly" means having general knowledge that a machine, instrument, tool, item of equipment, contrivance, or device is a drug related object or having reasonable grounds to believe that any such object is or may, to an average person, appear to be a drug related object. -

NEW ZEALAND DATA SHEET 1. PRODUCT NAME IOPIDINE® (Apraclonidine Hydrochloride) Eye Drops 0.5%
NEW ZEALAND DATA SHEET 1. PRODUCT NAME IOPIDINE® (apraclonidine hydrochloride) Eye Drops 0.5%. 2. QUALITATIVE AND QUANTITATIVE COMPOSITION Each mL of Iopidine Eye Drops 0.5% contains apraclonidine hydrochloride 5.75 mg, equivalent to apraclonidine base 5 mg. Excipient with known effect Benzalkonium chloride 0.1 mg per 1 mL as a preservative. For the full list of excipients, see section 6.1. 2. PHARMACEUTICAL FORM Eye drops, solution, sterile, isotonic. 4. CLINICAL PARTICULARS 4.1. Therapeutic indications Iopidine Eye Drops 0.5% are indicated for short-term adjunctive therapy in patients on maximally tolerated medical therapy who require additional IOP reduction. Patients on maximally tolerated medical therapy who are treated with Iopidine Eye Drops 0.5% to delay surgery should have frequent follow up examinations and treatment should be discontinued if the intraocular pressure rises significantly. The addition of Iopidine Eye Drops 0.5% to patients already using two aqueous suppressing drugs (i.e. beta-blocker plus carbonic anhydrase inhibitor) as part of their maximally tolerated medical therapy may not provide additional benefit. This is because apraclonidine is an aqueous-suppressing drug and the addition of a third aqueous suppressant may not significantly reduce IOP. The IOP-lowering efficacy of Iopidine Eye Drops 0.5% diminishes over time in some patients. This loss of effect, or tachyphylaxis, appears to be an individual occurrence with a variable time of onset and should be closely monitored. The benefit for most patients is less than one month. 4.2. Dose and method of administration Dose One drop of Iopidine Eye Drops 0.5% should be instilled into the affected eye(s) three times per day. -

ALPHAGAN® 0.2% Safely and Effectively
NDA 20613/S-031 Page 4 HIGHLIGHTS OF PRESCRIBING INFORMATION ___________ ___________ ADVERSE REACTIONS Most common adverse reactions occurring in These highlights do not include all the information needed approximately 10 to 30% of patients receiving brimonidine to use ALPHAGAN® 0.2% safely and effectively. See full ophthalmic solution 0.2% included oral dryness, ocular prescribing information for ALPHAGAN® 0.2%. hyperemia, burning and stinging, headache, blurring, foreign body sensation, fatigue/drowsiness, conjunctival ALPHAGAN® (brimonidine tartrate ophthalmic solution) follicles, ocular allergic reactions, and ocular pruritus. (6.1) 0.2% For Topical Ophthalmic Use To report SUSPECTED ADVERSE REACTIONS, Initial U.S. Approval: 1996 contact Allergan at 1-800-433-8871 or the FDA at 1 800-FDA-1088 or www.fda.gov/medwatch. _________ _________ INDICATIONS AND USAGE ___________ ____________ DRUG INTERACTIONS ALPHAGAN® 0.2% is an alpha adrenergic agonist indicated Antihypertensives/cardiac glycosides may lower blood for lowering intraocular pressure (IOP) in patients with open- pressure. (7.1) angle glaucoma or ocular hypertension. (1) ________ ________ Use with CNS depressants may result in an additive or DOSAGE AND ADMINISTRATION potentiating effect. (7.2) One drop in the affected eye(s), three times daily, Tricyclic antidepressants may potentially blunt the approximately 8 hours apart. (2) hypotensive effect of systemic clonidine. (7.3) _______ _______ DOSAGE FORMS AND STRENGTHS Monoamine oxidase inhibitors may result in increased Solution containing 2 mg/mL brimonidine tartrate. (3) hypotension. (7.4) _________ _____________ _______ ______ CONTRAINDICATIONS USE IN SPECIFIC POPULATIONS Neonates and infants (under the age of 2 years). (4.1) Use with caution in children > 2 years of age. -

New Medicines Committee Briefing March 2015 Simbrinza
New Medicines Committee Briefing March 2015 Simbrinza® (Brinzolamide 10mg + Brimonidine tartrate 2mg/ml) for treatment of elevated intraocular pressure (IOP) in adult patients with open-angle glaucoma or ocular hypertension. Simbrinza® is to be reviewed for use within: Primary Care Secondary Care Summary: Simbrinza® is a combination of brinzolamide which is a carbonic anhydrase (CA-II) inhibitor and brimonidine which is an alpha-2 adrenergic agonist. Simbrinza® is licensed for the treatment of elevated IOP in adult patients with open-angle glaucoma or ocular hypertension whom failed on mono-therapy. Simbrinza® is administered twice daily in the affected eye(s). Simbrinza® is the first combination therapy which does not contain a beta-blocker. SMC has accepted Simbrinza® for use within NHS Scotland, as there is no significant additional cost associated with the combination product compared with its individual components. One trial indicated that fixed combination of Simbrinza® administered BD had a significantly greater IOP lowering effect compared to either brinzolamide 1% or brimonidine 0.2% alone and displayed a safety profile consistent with its individual components. Another trial noted Simbrinza® to be non-inferior to separate administration of brimonidine and brinzolamide less than 10 min apart to prevent wash out. 1 Formulary application: Opthamology department: Consultant submitting application: Mr Lynval Jones (Consultant Ophthalmologist, Glaucoma) Clinical Director supporting application: Gareth Rowland (Clinical Director of Specialised Surgery) Mr Jones has requested for Simbrinza® to be considered for inclusion in the North Staffordshire Joint Formulary for the treatment of elevated intraocular pressure (IOP) in adult patients with open-angle glaucoma or ocular hypertension who have not responded to monotherapy. -

View Board, Austin, Texas, As Well As the Ethics Com- Pendent Examiner in the Same Room with the Same Instru- Mittee of Eye Center and Research, Aseer, Saudi Arabia
Abdelkader and Kaufman Eye and Vision (2016) 3:31 DOI 10.1186/s40662-016-0065-3 RESEARCH Open Access Clinical outcomes of combined versus separate carbachol and brimonidine drops in correcting presbyopia Almamoun Abdelkader1* and Herbert E. Kaufman2 Abstract Background: To test and compare in a masked fashion the efficacy of using a parasympathomimetic drug (3% carbachol) and an alpha-2 agonist (0.2% brimonidine) in both combined and separate forms to create optically beneficial miosis to pharmacologically improve vision in presbyopia. Methods: A prospective, double-masked, randomized, controlled clinical trial was conducted. Ten naturally emmetropic and presbyopic subjects between 42 and 58 years old with uncorrected distance visual acuity of at least 20/20 in both eyes without additional ocular pathology were eligible for inclusion. All subjects received 3% carbachol and 0.2% brimonidine in both combined and separate forms, 3% carbachol alone and 0.2% brimonidine (control) alone in their non-dominant eye in a crossover manner with one week washout between tests. The subjects’ pupil sizes and both near and distance visual acuities will be evaluated pre- and post-treatment at 1, 2, 4, and 8 h, by a masked examiner at the same room illumination. Results: Statistically significant improvement in mean near visual acuity (NVA) was achieved in all subjects who received combined 3% carbachol and 0.2% brimonidine in the same formula compared with those who received separate forms or carbachol alone or brimonidine alone (P < 0.0001). Conclusion: Based on the data, the combined solution demonstrated greater efficacy than the other solutions that were tested. -
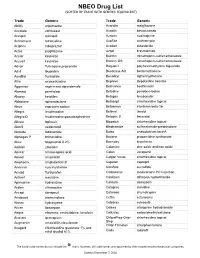
Drug List (SORTED by TRADE with GENERIC EQUIVALENT)
NBEO Drug List (SORTED BY TRADE WITH GENERIC EQUIVALENT) Trade Generic Trade Generic Abilify aripiprazole Avandia rosiglitazone Accolate zafirlukast Avastin bevacizumab Accupril quinapril Azasan azathioprine Achromycin tetracycline AzaSite azithromycin Aciphex rabeprazole Avodart dutasteride Actos pioglitazone Azopt brinzolamide Acular ketorolac Bactrim trimethoprim-sulfamethoxazole Acuvail ketorolac Bactrim DS trimethoprim-sulfamethoxazole Advair fluticasone propionate Baquacil polyhexamethylene biguanide Advil ibuprofen Beconase AQ beclomethasone AeroBid flunisolide Benadryl diphenhydramine Afrin oxymetazoline Bepreve Bepotastine besilate Aggrenox aspirin and dipyridamole Besivance besifloxacin Alamast pemirolast Betadine povidone-iodine Alaway ketotifen Betagan levobunolol Aldactone spironolactone Betasept chlorhexidine topical Aleve naproxen sodium Betaseron interferon beta-1b Allegra fexofenadine Betimol timolol Allegra-D fexofenadine-pseudoephedrine Betoptic S betaxolol Alluvia lopinavir Biopatch chlorhexidine topical Alocril nedocromil Blephamide sulfacetamide-prednisolone Alomide lodoxamide Botox onabotulinum toxinA Alphagan P brimonidine Brolene propamidine isethionate Alrex loteprednol 0.2% Bromday bromfenac Ambien zolpidem Calamine zinc oxide and iron oxide Amicar aminocaproic acid Calan verapamil Amoxil amoxicillin Calgon Vesta chlorhexidine topical Amphocin amphotericin B Capoten captopril Anectine succinylcholine Carafate sucralfate Ansaid flurbiprofen Carbocaine mepivacaine HCl injection Antivert meclizine Cardizem diltiazem -

IOPIDINE® 0.5% (Apraclonidine Ophthalmic Solution) 0.5% As Base
IOPIDINE® 0.5% (apraclonidine ophthalmic solution) 0.5% As Base DESCRIPTION: IOPIDINE® 0.5% Ophthalmic Solution contains apraclonidine hydrochloride, an alpha adrenergic agonist, in a sterile isotonic solution for topical application to the eye. Apraclonidine hydrochloride is a white to off-white powder and is highly soluble in water. Its chemical name is 2-[(4-amino-2,6 dichlorophenyl) imino]imidazolidine monohydrochloride with an empirical formula of C9H11Cl3N4 and a molecular weight of 281.57. The chemical structure of apraclonidine hydrochloride is: Each mL of IOPIDINE 0.5% Ophthalmic Solution contains: Active: apraclonidine hydrochloride 5.75 mg equivalent to apraclonidine base 5 mg; . Inactives: sodium chloride, sodium acetate, sodium hydroxide and/or hydrochloric acid (pH 4.4-7.8), purified water and benzalkonium chloride 0.01% (preservative) CLINICAL PHARMACOLOGY: Apraclonidine hydrochloride is a relatively selective alpha- 2-adrenergic agonist. When instilled in the eye, IOPIDINE 0.5% Ophthalmic Solution, has the action of reducing elevated, as well as normal, intraocular pressure (IOP), whether or not accompanied by glaucoma. Ophthalmic apraclonidine has minimal effect on cardiovascular parameters. Elevated IOP presents a major risk factor in glaucomatous field loss. The higher the level of IOP, the greater the likelihood of optic nerve damage and visual field loss. IOPIDINE 0.5% Ophthalmic Solution has the action of reducing IOP. The onset of action of apraclonidine can usually be noted within one hour, and maximum IOP reduction occurs about three hours after instillation. Aqueous fluorophotometry studies demonstrate that apraclonidine's predominant mechanism of action is reduction of aqueous flow via stimulation of the alpha-adrenergic system. -

Sustained-Release Compositions Containing Cation Exchange Resins and Polycarboxylic Polymers
~" ' Nil II II II II Ml Ml INI MINI II J European Patent Office *»%r\ n » © Publication number: 0 429 732 B1 Office_„. europeen des brevets © EUROPEAN PATENT SPECIFICATION © Date of publication of patent specification: 16.03.94 © Int. CI.5: A61 K 9/06, A61 K 9/1 8, A61 K 47/32 © Application number: 89312590.6 @ Date of filing: 01.12.89 © Sustained-release compositions containing cation exchange resins and polycarboxylic polymers. @ Date of publication of application: (73) Proprietor: ALCON LABORATORIES, INC. 05.06.91 Bulletin 91/23 6201 South Freeway Fort Worth Texas 76107(US) © Publication of the grant of the patent: 16.03.94 Bulletin 94/11 @ Inventor: Janl, Rajni 4621 Briarhaven Road © Designated Contracting States: Fort Worth, Texas 76109(US) AT BE CH DE FR GB IT LI LU NL SE Inventor: Harris, Robert Gregg 3224 Westcliff Road W. © References cited: Fort Worth, Texas 76109(US) EP-A- 0 254 822 J.Pharm. SCI., vol. 60, no. 9, September 1971, © Representative: Jump, Timothy John Simon et pages 1343-1345 A. HEYD:"Polymer-drug in- al teraction: stability of aqueous gels contain- Venner Shipley & Co. ing neomycin sulfate" 20 Little Britain London EC1A 7DH (GB) 00 CM CO o> CM Note: Within nine months from the publication of the mention of the grant of the European patent, any person ® may give notice to the European Patent Office of opposition to the European patent granted. Notice of opposition CL shall be filed in a written reasoned statement. It shall not be deemed to have been filed until the opposition fee LU has been paid (Art. -

Generic Drugs and Trade Name Equivalents: Alphabetized by Generic Name
GENERIC DRUGS AND TRADE NAME EQUIVALENTS: ALPHABETIZED BY GENERIC NAME Generic Trade Generic Trade Generic Trade acetaminophen Tylenol escitalopram Lexapro niacin, nicotinic acid Niaspan, Slo-Niacin acetaminophen-codeine Tylenol #3 estrogen Premarin nitroglycerin Nitro-bid acetazolamide Diamox eszopiclone Lunesta nystatin Mycostatin acyclovir Zovirax ethambutol Myambutol ofloxacin Ocuflox albuterol Proventil, Ventolin eye vitamin supplement Ocuvite PreserVision olopatadine Patanol alendronate Fosamax famciclovir Famvir omeprazole Prilosec allopurinol Zyloprim felodipine Plendil paraffin-mineral oil Lacri-Lube alprazolam Xanax fexofenadine Allegra paroxetine Paxil aminocaproic acid Amicar fexofenadine-pseudoephedrine Allegra-D pegaptanib Macugen amiodarone Cordarone finasteride Propecia, Proscar peginterferon alfa-2a Pegasys amitryptyline Elavil fluconazole Diflucan peginterferon alfa-2b PegIntron amlodipine Norvasc flunisolide AeroBid pemirolast potassium Alamast amoxicillin Amoxil, Dispermox fluorometholone FML phenytoin Dilantin amoxicillin-clavulanate Augmentin fluorouracil Efudex pilocarpine IsoptoCarpine, Pilocar apraclonidine Iopidine fluoxetine Prozac piroxicam Feldene AREDS formula PreserVision flutamide Eulexin polymyxin B-bacitracin Polysporin atenolol Tenormin fluticasone Flovent, Flonase polymyxin B-bacitracin-neomycin Neosporin (ointment) atorvastatin Lipitor folinic acid Folixor polymyxin B-neomycin-gramicidin Neosporin (suspension) azathioprine Imuran foscarnet Foscavir potassium chloride Micro-K, K-Dur, Klor-Com azelastine -

COMBIGAN® EYE DROPS (Brimonidine Tartrate 2.0 Mg Per Ml and Timolol (As Maleate) 5.0 Mg Per Ml)
COMBIGAN® EYE DROPS (brimonidine tartrate 2.0 mg per mL and timolol (as maleate) 5.0 mg per mL) Consumer Medicine Information What is in this leaflet aware of this gradual loss of sight. any of the ingredients listed at Sometimes even normal eye the end of this leaflet, some This leaflet answers some common pressure is associated with damage symptoms of an allergic reaction questions about COMBIGAN® eye to the back of the eye. include skin rash, itching, drops. It does not contain all the There are usually no symptoms of shortness of breath or swelling available information. It does not glaucoma. The only way of of the face, lips or tongue, which take the place of talking to your knowing that you have glaucoma is may cause difficulty in doctor or pharmacist. to have your eye pressure, optic swallowing or breathing All medicines have risks and nerve and visual field checked by an • you are taking monoamine benefits. Your doctor has weighed eye specialist or optometrist. If oxidase antidepressant the risks of you using glaucoma is not treated it can lead medication COMBIGAN® eye drops against to serious problems, including total • you have bronchospasm, the benefits they expect it will have blindness. In fact, untreated bronchial asthma or have a for you. glaucoma is one of the most history of bronchial asthma or common causes of blindness. other lung disease If you have any concerns about using this medicine, ask your doctor COMBIGAN® eye drops lower the • you have a severe or unstable or or pharmacist. pressure in the eye by decreasing uncontrolled heart condition Keep this leaflet with the the fluid produced and helping the • the seal around the cap is broken medicine.