Mutational Landscape of Spontaneous Base Substitutions and Small Indels in Experimental Caenorhabditis Elegans Populations of Differing Size
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Frameshift Indels Generate Highly Immunogenic Tumor Neoantigens Tumor-Specifi C Neoantigens Are the Targets of T Cells in the Neoantigens
Published OnlineFirst July 21, 2017; DOI: 10.1158/2159-8290.CD-RW2017-135 RESEARCH WATCH Apoptosis Major finding: An NMR-based fragment Mechanism: BIF-44 binds to a deep hy- Impact: Allosteric BAX sensitization screen identified a BAX-interacting com- drophobic pocket to induce conformation may represent a therapeutic strategy pound, BIF-44, that enhances BAX activity . changes that sensitize BAX activation . to promote apoptosis of cancer cells . BAX CAN BE ALLOSTERICALLY SENSITIZED TO PROMOTE APOPTOSIS The proapoptotic BAX protein is comprised of BH3 motif of the BIM protein. BIF-44 bound nine α-helices (α1–α9) and is a critical regulator competitively to the same region as vMIA, in a of the mitochondrial apoptosis pathway. In the deep hydrophobic pocket formed by the junction conformationally inactive state, BAX is primar- of the α3–α4 and α5–α6 hairpins that normally ily cytosolic and can be activated by BH3-only maintain BAX in an inactive state. Binding of BIF- activator proteins, which bind to ab α6/α6 “trig- 44 induced a structural change that resulted in ger site” to induce a conformational change that allosteric mobilization of the α1–α2 loop, which activates BAX and promotes its oligomerization. is involved in BH3-mediated activation, and the Conversely, antiapoptotic BCL2 proteins or the cytomeg- BAX BH3 helix, which is involved in propagating BAX oli- alovirus vMIA protein can bind to and inhibit BAX. Efforts gomerization, resulting in sensitization of BAX activation. In to therapeutically enhance apoptosis have largely focused addition to identifying a BAX allosteric sensitization site and on inhibiting antiapoptotic proteins. -

Small Variants Frequently Asked Questions (FAQ) Updated September 2011
Small Variants Frequently Asked Questions (FAQ) Updated September 2011 Summary Information for each Genome .......................................................................................................... 3 How does Complete Genomics map reads and call variations? ........................................................................... 3 How do I assess the quality of a genome produced by Complete Genomics?................................................ 4 What is the difference between “Gross mapping yield” and “Both arms mapped yield” in the summary file? ............................................................................................................................................................................. 5 What are the definitions for Fully Called, Partially Called, Half-Called and No-Called?............................ 5 In the summary-[ASM-ID].tsv file, how is the number of homozygous SNPs calculated? ......................... 5 In the summary-[ASM-ID].tsv file, how is the number of heterozygous SNPs calculated? ....................... 5 In the summary-[ASM-ID].tsv file, how is the total number of SNPs calculated? .......................................... 5 In the summary-[ASM-ID].tsv file, what regions of the genome are included in the “exome”? .............. 6 In the summary-[ASM-ID].tsv file, how is the number of SNPs in the exome calculated? ......................... 6 In the summary-[ASM-ID].tsv file, how are variations in potentially redundant regions of the genome counted? ..................................................................................................................................................................... -

Indelible Markers the Recruitment of Modified Histones by the RITS Complex
RESEARCH HIGHLIGHTS IN BRIEF EPIGENETICS Argonaute slicing is required for heterochromatic silencing and spreading. HUMAN GENETICS Irvine, D. V. et al. Science 313, 1134–1137 (2006) It has been proposed that small interfering RNA (siRNA)- guided histone H3 dimethylation on lysine 9 (H3K9me2) might be caused by an interaction of siRNA with DNA and INDELible markers the recruitment of modified histones by the RITS complex. Alternatively, siRNAs might guide histone modification by Over 10 million unique SNPs, some comprised about 30% of the base-pairing with RNA. Working in fission yeast, Irvine et al. of which influence human traits and total. Another ~30% consisted of provide support for the second mechanism. They show that disease susceptibilities, have been expansions of either monomeric the endonucleolytic cleavage motif of Argonaute is required identified in the human genome. base-pair repeats or multi-base for heterochromatic silencing and for ‘slicing’ mRNAs that are Now, another type of natural genetic repeats. Approximately 40% of indels complementary to siRNAs. They also show that spreading of variation, which involves insertion included insertions of apparently ran- silencing requires read-through transcription, as well as slicing. and deletion polymorphisms (indels), dom DNA sequences. Transposons has been systematically studied and accounted for only a small proportion TECHNOLOGY mapped for the first time. (less than 1%) of the polymorphisms Trans-kingdom transposition of the maize Dissociation Understanding more about indels that were identified. element. is important because they are known Indels were spread throughout Emelyanov, A. et al. Genetics 1 September 2006 (doi:10.1534/ to contribute to human disease. -

Pervasive Indels and Their Evolutionary Dynamics After The
MBE Advance Access published April 24, 2012 Pervasive Indels and Their Evolutionary Dynamics after the Fish-Specific Genome Duplication Baocheng Guo,1,2 Ming Zou,3 and Andreas Wagner*,1,2 1Institute of Evolutionary Biology and Environmental Studies, University of Zurich, Zurich, Switzerland 2The Swiss Institute of Bioinformatics, Quartier Sorge-Batiment Genopode, Lausanne, Switzerland 3Key Laboratory of Aquatic Biodiversity and Conservation, Institute of Hydrobiology, Chinese Academy of Sciences, Wuhan, People’s Republic of China *Corresponding author: E-mail: [email protected]. Associate editor: Herve´ Philippe Abstract Research article Insertions and deletions (indels) in protein-coding genes are important sources of genetic variation. Their role in creating new proteins may be especially important after gene duplication. However, little is known about how indels affect the divergence of duplicate genes. We here study thousands of duplicate genes in five fish (teleost) species with completely sequenced genomes. The ancestor of these species has been subject to a fish-specific genome duplication (FSGD) event Downloaded from that occurred approximately 350 Ma. We find that duplicate genes contain at least 25% more indels than single-copy genes. These indels accumulated preferentially in the first 40 my after the FSGD. A lack of widespread asymmetric indel accumulation indicates that both members of a duplicate gene pair typically experience relaxed selection. Strikingly, we observe a 30–80% excess of deletions over insertions that is consistent for indels of various lengths and across the five genomes. We also find that indels preferentially accumulate inside loop regions of protein secondary structure and in http://mbe.oxfordjournals.org/ regions where amino acids are exposed to solvent. -

The Origin, Evolution, and Functional Impact of Short Insertion–Deletion Variants Identified in 179 Human Genomes
Downloaded from genome.cshlp.org on October 4, 2021 - Published by Cold Spring Harbor Laboratory Press Research The origin, evolution, and functional impact of short insertion–deletion variants identified in 179 human genomes Stephen B. Montgomery,1,2,3,14,16 David L. Goode,3,14,15 Erika Kvikstad,4,13,14 Cornelis A. Albers,5,6 Zhengdong D. Zhang,7 Xinmeng Jasmine Mu,8 Guruprasad Ananda,9 Bryan Howie,10 Konrad J. Karczewski,3 Kevin S. Smith,2 Vanessa Anaya,2 Rhea Richardson,2 Joe Davis,3 The 1000 Genomes Pilot Project Consortium, Daniel G. MacArthur,5,11 Arend Sidow,2,3 Laurent Duret,4 Mark Gerstein,8 Kateryna D. Makova,9 Jonathan Marchini,12 Gil McVean,12,13 and Gerton Lunter13,16 1–13[Author affiliations appear at the end of the paper.] Short insertions and deletions (indels) are the second most abundant form of human genetic variation, but our un- derstanding of their origins and functional effects lags behind that of other types of variants. Using population-scale sequencing, we have identified a high-quality set of 1.6 million indels from 179 individuals representing three diverse human populations. We show that rates of indel mutagenesis are highly heterogeneous, with 43%–48% of indels occurring in 4.03% of the genome, whereas in the remaining 96% their prevalence is 16 times lower than SNPs. Polymerase slippage can explain upwards of three-fourths of all indels, with the remainder being mostly simple de- letions in complex sequence. However, insertions do occur and are significantly associated with pseudo-palindromic sequence features compatible with the fork stalling and template switching (FoSTeS) mechanism more commonly as- sociated with large structural variations. -

The Genomics Era: the Future of Genetics in Medicine - Glossary
The Genomics Era: the Future of Genetics in Medicine - Glossary The glossary below provides a list of key terms used throughout the course. You do not need to read them all now; we’ll be linking back to the main glossary step wherever these terms appear, so you may refer back to this list if you are unsure of the terminology being used. Term Definition The process of matching reads back to their original Alignment position in the reference genome. An allele is one of a number of alternative forms of the same gene or genetic locus. We inherit one copy Allele of our genetic code from our mother and one copy of our genetic code from our father. Each copy is known as an allele. Microarray based genomic comparative hybridisation. This is a technique used to detect chromosome imbalances by comparing patient and control DNA and comparing differences between the two sets. It is Array CGH a useful technique for detecting small chromosome deletions and duplications which would not have been detected with more traditional karyotyping techniques. A unit of DNA. There are four bases which form the Base cross links (or rungs) of the DNA double helix: adenine (A), thymine (T), guanine (G) and cytosine (C). Capture see Target enrichment. The process by which a cell becomes specialized in Cell differentiation order to perform a specific function. Centromere The point at which the sister chromatids are joined. #1 FutureLearn A structure located in the nucleus all living cells, comprised of DNA bound around proteins called histones. The normal number of chromosomes in each Chromosome human cell nucleus is 46 and is composed of 22 pairs of autosomes and a pair of sex chromosomes which determine gender: males have an X and a Y chromosome whilst females have two X chromosomes. -

Integrated Analysis of Gene Expression, SNP, Indel, and CNV
G C A T T A C G G C A T genes Article Integrated Analysis of Gene Expression, SNP, InDel, and CNV Identifies Candidate Avirulence Genes in Australian Isolates of the Wheat Leaf Rust Pathogen Puccinia triticina Long Song , Jing Qin Wu, Chong Mei Dong and Robert F. Park * Plant Breeding Institute, School of Life and Environmental Science, Faculty of Science, The University of Sydney, Sydney 2006, NSW, Australia; [email protected] (L.S.); [email protected] (J.Q.W.); [email protected] (C.M.D.) * Correspondence: [email protected]; Tel.: +61-2-9351-8806; Fax: +61-2-9351-8875 Received: 3 September 2020; Accepted: 18 September 2020; Published: 21 September 2020 Abstract: The leaf rust pathogen, Puccinia triticina (Pt), threatens global wheat production. The deployment of leaf rust (Lr) resistance (R) genes in wheat varieties is often followed by the development of matching virulence in Pt due to presumed changes in avirulence (Avr) genes in Pt. Identifying such Avr genes is a crucial step to understand the mechanisms of wheat-rust interactions. This study is the first to develop and apply an integrated framework of gene expression, single nucleotide polymorphism (SNP), insertion/deletion (InDel), and copy number variation (CNV) analysis in a rust fungus and identify candidate avirulence genes. Using a long-read based de novo genome assembly of an isolate of Pt (‘Pt104’) as the reference, whole-genome resequencing data of 12 Pt pathotypes derived from three lineages Pt104, Pt53, and Pt76 were analyzed. Candidate avirulence genes were identified by correlating virulence profiles with small variants (SNP and InDel) and CNV, and RNA-seq data of an additional three Pt isolates to validate expression of genes encoding secreted proteins (SPs). -

Comprehensive Analysis of Indels in Whole-Genome Microsatellite Regions and Microsatellite Instability Across 21 Cancer Types
Downloaded from genome.cshlp.org on October 3, 2021 - Published by Cold Spring Harbor Laboratory Press Research Comprehensive analysis of indels in whole-genome microsatellite regions and microsatellite instability across 21 cancer types Akihiro Fujimoto,1,2,3 Masashi Fujita,1 Takanori Hasegawa,4 Jing Hao Wong,2,3 Kazuhiro Maejima,1 Aya Oku-Sasaki,1 Kaoru Nakano,1 Yuichi Shiraishi,5,6 Satoru Miyano,4,6 Go Yamamoto,7 Kiwamu Akagi,7 Seiya Imoto,4 and Hidewaki Nakagawa1 1Laboratory for Cancer Genomics, RIKEN Center for Integrative Medical Sciences, Tokyo 230-0045, Japan; 2Department of Human Genetics, The University of Tokyo, Graduate School of Medicine, Tokyo 113-0033, Japan; 3Department of Drug Discovery Medicine, Kyoto University Graduate School of Medicine, Kyoto 606-8507, Japan; 4Health Intelligence Center, Institute of Medical Sciences, The University of Tokyo, Tokyo 108-8639, Japan; 5Division of Cellular Signaling, National Cancer Center Research Institute, Tokyo 104-0045, Japan; 6Human Genome Center, Institute of Medical Sciences, The University of Tokyo, Tokyo 108-8639, Japan; 7Division of Molecular Diagnosis and Cancer Prevention, Saitama Cancer Center, Saitama 362-0806, Japan Microsatellites are repeats of 1- to 6-bp units, and approximately 10 million microsatellites have been identified across the human genome. Microsatellites are vulnerable to DNA mismatch errors and have thus been used to detect cancers with mismatch repair deficiency. To reveal the mutational landscape of microsatellite repeat regions at the genome level, we an- alyzed approximately 20.1 billion microsatellites in 2717 whole genomes of pan-cancer samples across 21 tissue types. First, we developed a new insertion and deletion caller (MIMcall) that takes into consideration the error patterns of different types of microsatellites. -

Indel Information Eliminates Trivial Sequence Alignment in Maximum Likelihood Phylogenetic Analysis
Cladistics Cladistics 28 (2012) 514–528 10.1111/j.1096-0031.2012.00402.x Indel information eliminates trivial sequence alignment in maximum likelihood phylogenetic analysis John S.S. Dentona,b,* and Ward C. Wheelerb,c aDivision of Vertebrate Zoology, American Museum of Natural History, New York, NY 10024, USA; bRichard Gilder Graduate School, American Museum of Natural History, New York, NY 10024, USA; cDivision of Invertebrate Zoology, American Museum of Natural History, New York, NY 10024, USA Accepted 12 March 2012 Abstract Although there has been a recent proliferation in maximum-likelihood (ML)-based tree estimation methods based on a fixed sequence alignment (MSA), little research has been done on incorporating indel information in this traditional framework. We show, using a simple model on a single character example, that a trivial alignment of a different form than that previously identified for parsimony is optimal in ML under standard assumptions treating indels as ‘‘missing’’ data, but that it is not optimal when indels are incorporated into the character alphabet. We show that the optimality of the trivial alignment is not an artefact of simplified theory assumptions by demonstrating that trivial alignment likelihoods of five different multiple sequence alignment datasets exhibit this phenomenon. These results demonstrate the need for use of indel information in likelihood analysis on fixed MSAs, and suggest that caution must be exercised when drawing conclusions from software implementations claiming improvements in likelihood scores under an indels-as-missing assumption. Ó The Willi Hennig Society 2012. Maximum likelihood (ML) has become a popular the increasing sophistication of search heuristics in ML optimality criterion for inferring evolutionary trees software (e.g. -

Accurate Detection and Classification of Heterozygous Indels by Direct Sequencing / 1550/F4
Accurate detection and classification of heterozygous indels by direct sequencing / 1550/F4 Jon Sorenson, Anjali Pradhan, Sharada Vijaychander, Bimal Sangari, Sylvia Fang, Theresa Nguyen, Ben Jones, Danwei Guo, Quynh Doan. Applied Biosystems, 850 Lincoln Centre Dr., Foster City, CA ABSTRACT Examples of heterozygous indels found by Algorithm results direct sequencing A heterozygous insertion/deletion (indel) mutation is defined by the presence of two alleles differing by an insertion or deletion---a length polymorphism. It has been estimated that 20% of the Figure 3a. polymorphisms in the human genome are length polymorphisms. Deletion of However the direct sequencing of heterozygous individuals is complicated by a “phase shift” in the electropherogram trace at the T near exon site of the indel mutation. Quality-based sequencing pipelines 2 of MSX1 discard this data as noise or spend significant time and effort trying to call the polymorphism correctly. Starting with SeqScape® (LL:4487). Software v2.0, we introduced an algorithm to predict when samples Table I. Testing results for the HIM detection algorithm implemented in were exhibiting the presence of a heterozygous indel mutation SeqScape Software v2.5. Results are grouped by the quality of the (HIM). This algorithm has been refined in subsequent releases of underlying data with respect to blobs, n+1 primer peaks, PCR the software to include basecalling the inserted/deleted sequence, contamination. Sensitivity is measured with two definitions: 1. How mapping the polymorphism to the reference sequence, displaying often was the true location and sequence called correctly? 2. How often the mutation in the HUGO-approved format, and assigning a quality Figure was the true location called correctly? As can be seen from these value to the mutation. -

CRISPR-Cas9 DNA Base-Editing and Prime-Editing
International Journal of Molecular Sciences Review CRISPR-Cas9 DNA Base-Editing and Prime-Editing Ariel Kantor 1,2,*, Michelle E. McClements 1,2 and Robert E. MacLaren 1,2 1 Nuffield Laboratory of Ophthalmology, Nuffield Department of Clinical Neurosciences & NIHR Oxford Biomedical Research Centre, University of Oxford, Oxford OX3 9DU, UK 2 Oxford Eye Hospital, Oxford University Hospitals NHS Foundation Trust, Oxford OX3 9DU, UK * Correspondence: [email protected] Received: 14 July 2020; Accepted: 25 August 2020; Published: 28 August 2020 Abstract: Many genetic diseases and undesirable traits are due to base-pair alterations in genomic DNA. Base-editing, the newest evolution of clustered regularly interspaced short palindromic repeats (CRISPR)-Cas-based technologies, can directly install point-mutations in cellular DNA without inducing a double-strand DNA break (DSB). Two classes of DNA base-editors have been described thus far, cytosine base-editors (CBEs) and adenine base-editors (ABEs). Recently, prime-editing (PE) has further expanded the CRISPR-base-edit toolkit to all twelve possible transition and transversion mutations, as well as small insertion or deletion mutations. Safe and efficient delivery of editing systems to target cells is one of the most paramount and challenging components for the therapeutic success of BEs. Due to its broad tropism, well-studied serotypes, and reduced immunogenicity, adeno-associated vector (AAV) has emerged as the leading platform for viral delivery of genome editing agents, including DNA-base-editors. In this review, we describe the development of various base-editors, assess their technical advantages and limitations, and discuss their therapeutic potential to treat debilitating human diseases. -
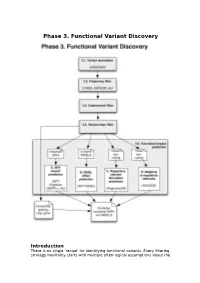
Phase 3. Functional Variant Discovery
Phase 3. Functional Variant Discovery Introduction There is no single ‘recipe’ for identifying functional variants. Every filtering strategy inevitably starts with multiple often-logical assumptions about the properties of causal variants that may also discard candidates too early. Novelty is one example, where the variant is not expected to be present in public repositories, even though apparently healthy individuals carrying it may have been sequenced in unrelated studies. High predicted functional impact is another, where a variant is expected to simultaneously occur at an evolutionarily conserved genomic location and also be scored as deleterious by multiple predictive algorithms may even be too stringent a rule in a Mendelian trait. In a similar vein, in a population genetics study, a novel amino acid class-changing variant that is not predicted to have a functional effect by the standard tools may have a molecular effect. An inheritance model may itself be incorrect, e.g. a condition may be autosomal recessive rather than X-linked as hypothesized, or digenic rather than recessive, etc. It is therefore important not to conclude too early that the variant(s) of interest “are not in the exome” and to exhaust all alternate plausible rules and scenarios before that conclusion is reached. Using disease variant discovery as a context, we aim to present here a range of guidelines that can be used to develop an appropriate variant prioritisation strategy. This phase will be presented as 5 sub-phases: 1. Variant annotation - using ANNOVAR (Wang et al, 2010) as an example 2. Frequency 3. Case-control filtering 4. Variant class filtering 5.