Case Report: Eruptive Xanthoma in a 14-Year-Old Boy Ryan M
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Apoa5genetic Variants Are Markers for Classic Hyperlipoproteinemia
CLINICAL RESEARCH CLINICAL RESEARCH www.nature.com/clinicalpractice/cardio APOA5 genetic variants are markers for classic hyperlipoproteinemia phenotypes and hypertriglyceridemia 1 1 1 2 2 1 1 Jian Wang , Matthew R Ban , Brooke A Kennedy , Sonia Anand , Salim Yusuf , Murray W Huff , Rebecca L Pollex and Robert A Hegele1* SUMMARY INTRODUCTION Hypertriglyceridemia is a common biochemical Background Several known candidate gene variants are useful markers for diagnosing hyperlipoproteinemia. In an attempt to identify phenotype that is observed in up to 5% of adults. other useful variants, we evaluated the association of two common A plasma triglyceride concentration above APOA5 single-nucleotide polymorphisms across the range of classic 1.7 mmol/l is a defining component of the meta 1 hyperlipoproteinemia phenotypes. bolic syndrome and is associated with several comorbidities, including increased risk of cardio Methods We assessed plasma lipoprotein profiles and APOA5 S19W and vascular disease2 and pancreatitis.3,4 Factors, –1131T>C genotypes in 678 adults from a single tertiary referral lipid such as an imbalance between caloric intake and clinic and in 373 normolipidemic controls matched for age and sex, all of expenditure, excessive alcohol intake, diabetes, European ancestry. and use of certain medications, are associated Results We observed significant stepwise relationships between APOA5 with hypertriglyceridemia; however, genetic minor allele carrier frequencies and plasma triglyceride quartiles. The factors are also important.5,6 odds ratios for hyperlipoproteinemia types 2B, 3, 4 and 5 in APOA5 S19W Complex traits, such as plasma triglyceride carriers were 3.11 (95% CI 1.63−5.95), 4.76 (2.25−10.1), 2.89 (1.17−7.18) levels, usually do not follow Mendelian patterns of and 6.16 (3.66−10.3), respectively. -

The Effect of Omega-3 Fatty Acids on Hypertriglyceridemia: a Review
Review Article ISSN: 2574 -1241 DOI: 10.26717/BJSTR.2020.26.004382 The Effect of Omega-3 Fatty Acids on Hypertriglyceridemia: A Review Alaa Abousetta, Ibrahim Abousetta, Theresa Dobler, Lia Ebrahimi, Vassillis Frangoullis, Stephanos Christodoulides and Ioannis Patrikios* School of Medicine, European University Cyprus, Cyprus *Corresponding author: Ioannis Patrikios, School of Medicine, European University Cyprus, Cyprus ARTICLE INFO Abstract Received: March 06, 2020 Hypertriglyceridemia is a common problem in adults in the developed world. It is associated with increased levels of triglycerides within the blood, which subsequently Published: March 17, 2020 promote the development of other diseases such as cardiovascular disease and pancreatitis. Triglycerides are mostly consumed through the diet and act as a source of energy in between meals. However, the levels of triglycerides increase proportionally Citation: Alaa A, Ibrahim A, Theresa D, Lia with the number of calories consumed. This only leads to a problem if the total daily E, Ioannis P, et al., The Effect of Omega-3 energy expenditure is exceeded. Additionally, the type of macronutrients taken in Fatty Acids on Hypertriglyceridemia: A Re- view. Biomed J Sci & Tech Res 26(4)-2020. for instance, high carbohydrate intake has been associated with an increased plasma BJSTR. MS.ID.004382. triglyceridethrough the level.diet has If thea direct levels influence of the triglycerideson having an inincreased the blood level rise of abovetriglycerides, 150 mg/ as Abbreviations: FHTG: Familial hypertri- dL, it is considered as hypertriglyceridemia. Increasing numbers of triglycerides can glyceridemia; FCH: Familial Combined Hy- have multiple underlying causes, which can be divided into primary and secondary perlipidemia, systemic lupus erythemato- causes. -

Other Types of Primary Hyperlipoproteinemia
82 Journal of Atherosclerosis and Thrombosis Vol.21, No.2 Committee Report 10 Other Types of Primary Hyperlipoproteinemia (Hyperlipidemia) Executive Summary of the Japan Atherosclerosis Society (JAS) Guidelines for the Diagnosis and Prevention of Atherosclerotic Cardiovascular Diseases in Japan ― 2012 Version Tamio Teramoto, Jun Sasaki, Shun Ishibashi, Sadatoshi Birou, Hiroyuki Daida, Seitaro Dohi, Genshi Egusa, Takafumi Hiro, Kazuhiko Hirobe, Mami Iida, Shinji Kihara, Makoto Kinoshita, Chizuko Maruyama, Takao Ohta, Tomonori Okamura, Shizuya Yamashita, Masayuki Yokode and Koutaro Yokote Committee for Epidemiology and Clinical Management of Atherosclerosis 1. Primary Hyperlipoproteinemias (Hyperlipidemias) enhanced hepatic apolipoprotein (apo) B-100 synthe- Other Than Familial Hypercholesterolemia sis, decreased LPL activity, increased very-low-density There are various types of primary hyperlipopro- lipoprotein (VLDL) secretion from the liver and the teinemias (hyperlipidemias) other than familial hyper- accumulation of visceral fat as factors for the develop- cholesterolemia (FH). These types are clinically ment of symptoms and has been reported to be related important and classified according to their associated to abnormalities of the LPL and APOC-Ⅱ genes or pathophysiology and genetic abnormalities (Table 1)1). APOA-Ⅰ/C-Ⅲ/A-Ⅳ gene cluster. However, none of Familial lipoprotein lipase (LPL) deficiency manifests these findings have been proven to be definitive. It has as severe hyperchylomicronemia and may present with also been suggested that FCHL is caused by a poly- eruptive cutaneous xanthomas or acute pancreatitis, genic background that tends to induce hyperlipopro- although it does not necessarily accompany atheroscle- teinemia due to environmental factors, such as over- rotic cardiovascular disease (CVD). On the other nutrition and a low level of physical activity. -

Severe Hypertriglyceridemia in a Patient Heterozygous for a Lipoprotein Lipase Gene Allele with Two Novel Missense Variants
European Journal of Human Genetics (2015) 23, 1259–1261 & 2015 Macmillan Publishers Limited All rights reserved 1018-4813/15 www.nature.com/ejhg SHORT REPORT Severe hypertriglyceridemia in a patient heterozygous for a lipoprotein lipase gene allele with two novel missense variants Ursula Kassner*,1, Bastian Salewsky2,3, Marion Wühle-Demuth1, Istvan Andras Szijarto1, Thomas Grenkowitz1, Priska Binner4, Winfried März4,5,6, Elisabeth Steinhagen-Thiessen1,2 and Ilja Demuth*,2,3 Rare monogenic hyperchylomicronemia is caused by loss-of-function mutations in genes involved in the catabolism of triglyceride-rich lipoproteins, including the lipoprotein lipase gene, LPL. Clinical hallmarks of this condition are eruptive xanthomas, recurrent pancreatitis and abdominal pain. Patients with LPL deficiency and severe or recurrent pancreatitis are eligible for the first gene therapy treatment approved by the European Union. Therefore the precise molecular diagnosis of familial hyperchylomicronemia may affect treatment decisions. We present a 57-year-old male patient with excessive hypertriglyceridemia despite intensive lipid-lowering therapy. Abdominal sonography showed signs of chronic pancreatitis. Direct DNA sequencing and cloning revealed two novel missense variants, c.1302A4T and c.1306G4A, in exon 8 of the LPL gene coexisting on the same allele. The variants result in the amino-acid exchanges p.(Lys434Asn) and p.(Gly436Arg). They are located in the carboxy-terminal domain of lipoprotein lipase that interacts with the glycosylphosphatidylinositol- anchored HDL-binding protein (GPIHBP1) and are likely of functional relevance. No further relevant mutations were found by direct sequencing of the genes for APOA5, APOC2, LMF1 and GPIHBP1. We conclude that heterozygosity for damaging mutations of LPL may be sufficient to produce severe hypertriglyceridemia and that chylomicronemia may be transmitted in a dominant manner, at least in some families. -

Evaluation and Treatment of Hypertriglyceridemia: an Endocrine Society Clinical Practice Guideline
SPECIAL FEATURE Clinical Practice Guideline Evaluation and Treatment of Hypertriglyceridemia: An Endocrine Society Clinical Practice Guideline Lars Berglund, John D. Brunzell, Anne C. Goldberg, Ira J. Goldberg, Frank Sacks, Mohammad Hassan Murad, and Anton F. H. Stalenhoef University of California, Davis (L.B.), Sacramento, California 95817; University of Washington (J.D.B.), Seattle, Washington 98195; Washington University School of Medicine (A.C.G.), St. Louis, Missouri 63110; Columbia University (I.J.G.), New York, New York 10027; Harvard School of Public Health (F.S.), Boston, Massachusetts 02115; Mayo Clinic (M.H.M.), Rochester, Minnesota 55905; and Radboud University Nijmegen Medical Centre (A.F.H.S.), 6525 GA Nijmegen, The Netherlands Objective: The aim was to develop clinical practice guidelines on hypertriglyceridemia. Participants: The Task Force included a chair selected by The Endocrine Society Clinical Guidelines Subcommittee (CGS), five additional experts in the field, and a methodologist. The authors received no corporate funding or remuneration. Consensus Process: Consensus was guided by systematic reviews of evidence, e-mail discussion, conference calls, and one in-person meeting. The guidelines were reviewed and approved sequen- tially by The Endocrine Society’s CGS and Clinical Affairs Core Committee, members responding to a web posting, and The Endocrine Society Council. At each stage, the Task Force incorporated changes in response to written comments. Conclusions: The Task Force recommends that the diagnosis of hypertriglyceridemia be based on fasting levels, that mild and moderate hypertriglyceridemia (triglycerides of 150–999 mg/dl) be diagnosed to aid in the evaluation of cardiovascular risk, and that severe and very severe hyper- triglyceridemia (triglycerides of Ͼ 1000 mg/dl) be considered a risk for pancreatitis. -

Familial Partial Lipodystrophy
Familial Partial Lipodystrophy Purvisha Patel; Ralph Starkey, MD; Michele Maroon, MD The lipodystrophies are rare disorders character- ized by insulin resistance and the absence or loss of body fat. The 4 subtypes of lipodystrophy are characterized by onset and distribution. Partial lipodystrophy is rare, with loss of fat from the extremities and excess fat accumulation in the face and neck; recognizing this phenotype and subsequent referral for endocrinologic care may improve outcome and reduce mortality. ipodystrophies are rare disorders characterized by insulin resistance and the absence or L loss of body fat.1 Classification of the 4 main subtypes of lipodystrophy is based on onset (congenital/familial vs acquired/sporadic) and dis- Figure not available online tribution (total/generalized vs partial). Congenital total lipodystrophy (also known as Berardinelli syndrome, Seip syndrome) is a rare autosomal- recessive disorder marked by an almost complete lack of adipose tissue from birth. Familial partial lipodystrophy (also known as Kobberling-Dunnigan syndrome) involves loss of subcutaneous fat from the extremities and accumulation of excess fat in the face and neck and to a lesser extent in the hands and feet. Acquired total lipodystrophy (also known as lipoatrophy, Lawrence-Seip syndrome) presents with generalized loss of fat beginning in childhood. Acquired partial lipodystrophy (also known as progressive lipodystrophy, partial lipoat- Figure 1. Accentuation of fat pads in the face and neck. rophy, Barraquer-Simons syndrome) is character- ized by loss of fat only from the upper extremities, face, and trunk.2 tional uterine bleeding (gravida 2, para 1, AB 1) Case Report necessitating total hysterectomy. On physical A 39-year-old white woman presented with the examination, accentuation of fat pads in the face complaint of thickened brown skin on the neck and and neck (Figure 1), central obesity, and prominent medial thighs. -

Management of Hypertriglyceridemia BMJ: First Published As 10.1136/Bmj.M3109 on 12 October 2020
STATE OF THE ART REVIEW Management of hypertriglyceridemia BMJ: first published as 10.1136/bmj.m3109 on 12 October 2020. Downloaded from Vinaya Simha ABSTRACT Hypertriglyceridemia is one of the most common lipid abnormalities encountered in clinical practice. Many monogenic disorders causing severe hypertriglyceridemia have been identified, but in most patients triglyceride elevations result from a Division of Endocrinology, Mayo combination of multiple genetic variations with small effects and environmental Clinic, Rochester, MN 55905, USA factors. Common secondary causes include obesity, uncontrolled diabetes, alcohol Correspondence to: misuse, and various commonly used drugs. Correcting these factors and optimizing [email protected] Cite this as: BMJ 2020;371:m3109 lifestyle choices, including dietary modification, is important before starting http://dx.doi.org/10.1136/bmj.m3109 drug treatment. The goal of drug treatment is to reduce the risk of pancreatitis in Series explanation: State of the Art Reviews are commissioned patients with severe hypertriglyceridemia and cardiovascular disease in those on the basis of their relevance with moderate hypertriglyceridemia. This review discusses the various genetic and to academics and specialists in the US and internationally. acquired causes of hypertriglyceridemia, as well as current management strategies. For this reason they are written predominantly by US authors. Evidence supporting the different drug and non-drug approaches to treating hypertriglyceridemia is examined, and an easy to adopt step-by-step management strategy is presented. Introduction independent risk factor for ASCVD and optimal drug Hypertriglyceridemia is a fairly common clinical treatment, including novel and emerging therapies, condition, but it continues to evoke considerable to mitigate this will be examined in greater detail. -

Familial Combined Hyperlipidemia: Current Knowledge, Perspectives
REVISTA DE INVESTIGACIÓN CLÍNICA Contents available at PubMed www .clinicalandtranslationalinvestigation .com PERMANYER Rev Invest Clin. 2018;70:224-36 BRIEF REVIEW Familial Combined Hyperlipidemia: Current Knowledge, Perspectives, and Controversies Omar Yaxmehen Bello-Chavolla1,2, Anuar Kuri-García1, Monserratte Ríos-Ríos1, Arsenio Vargas-Vázquez1,2, Jorge Eduardo Cortés-Arroyo1,2, Gabriela Tapia-González1, Ivette Cruz-Bautista1,3,4 and Carlos Alberto Aguilar-Salinas1,3,4* 1Metabolic Diseases Research Unit and 3Department of Endocrinology and Metabolism, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, Mexico City, Mexico; 2MD/PhD (PECEM) Program, Facultad de Medicina, Universidad Nacional Autónoma de México; 4Tec Salud, Instituto Tecnológico y de Estudios Superiores de Monterrey, Monterrey, N.L., México ABSTRACT Familial combined hyperlipidemia (FCHL) is the most prevalent primary dyslipidemia; however, it frequently remains undiagnosed and its precise definition is a subject of controversy. FCHL is characterized by fluctuations in serum lipid concentrations and may present as mixed hyperlipidemia, isolated hypercholesterolemia, hypertriglyceridemia, or as a normal serum lipid profile in combination with abnormally elevated levels of apolipoprotein B. FCHL is an oligogenic primary lipid disorder, which can occur due to the interaction of several contributing variants and mutations along with environmental triggers. Controversies surround- ing the relevance of identifying FCHL as a cause of isolated hypertriglyceridemia and a differential diagnosis of familial hyper- triglyceridemia are offset by the description of associations with USF1 and other genetic traits that are unique for FCHL and that are shared with other conditions with similar pathophysiological mechanisms. Patients with FCHL are at an increased risk of cardiovascular disease and mortality and have a high frequency of comorbidity with other metabolic conditions such as type 2 diabetes, non-alcoholic fatty liver disease, steatohepatitis, and the metabolic syndrome. -

Eruptive Xanthomas: a Warning Sign of Future Hyperlipidemia Complications
International Journal of Research in Dermatology Nessel TA et al. Int J Res Dermatol. 2020 Jul;6(4):579-583 http://www.ijord.com DOI: http://dx.doi.org/10.18203/issn.2455-4529.IntJResDermatol20202672 Case Report Eruptive xanthomas: a warning sign of future hyperlipidemia complications Trevor A. Nessel1*, Connor C. Kerndt2, John A. Bills1, Lynn Sikorski3 1Department of Dermatology, Michigan State University College of Osteopathic Medicine, East Lansing, Michigan, USA 2Department of Dermatology, Spectrum Health/Michigan State University College of Human Medicine, Grand Rapids, Michigan, USA 3Department of Dermatology, Beaumont Farmington Hills Hospital, Farmington Hills, Michigan, USA Received: 08 April 2020 Accepted: 07 May 2020 *Correspondence: Trevor A. Nessel, E-mail: [email protected] Copyright: © the author(s), publisher and licensee Medip Academy. This is an open-access article distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited. ABSTRACT Eruptive xanthomas are localized lipid deposits in the skin or subcutaneous tissue that are associated with both primary and secondary hyperlipidemia. Typical presentation manifests as small yellow papules on the buttocks or extensor surfaces. Xanthomas can be diagnosed clinically with an extensive history and physical examination, however, can be confirmed via biopsy and histological findings. It is essential to identify the underlying cause of the skin lesions and take appropriate measures to prevent future hyperlipidemia-induced consequences. Here we report a 42-year-old female with eruptive xanthomas on her trunk and extremities. Previous visits to the primary care provider and emergency department resulted in diagnoses of viral exanthems. -

Transient Dyslipidemia Mimicking the Plasma Lipid Profile of Tangier Disease in a Diabetic Patient with Gram Negative Sepsis
Available online at www.annclinlabsci.org 150 Annals of Clinical & Laboratory Science, vol. 41, no. 2, 2011 Transient Dyslipidemia Mimicking the Plasma Lipid Profile of Tangier Disease in a Diabetic Patient with Gram Negative Sepsis Carlos Palacio, M.D.,1 Irene Alexandraki, M.D.,1 Roger L. Bertholf, Ph.D.,2 and Arshag D. Mooradian, M.D.1 Department of Medicine1 and the Department of Pathology2, University of Florida - College of Medicine, Jacksonville, FL Abstract. Tangier disease is a rare genetic disorder of lipid metabolism characterized by low concentrations of plasma high density lipoprotein (HDL) and low density lipoprotein (LDL) cholesterol with normal or elevated levels of triglycerides. In this case report we describe a patient with diabetes who experienced an episode of urosepsis with a plasma lipid profile resembling Tangier disease. Experimental evidence in the literature suggests that similar lipid changes may occur due to cytokines released during sepsis. Clinicians should be aware of these changes to avoid misdiagnosis of lipid disorders. Introduction Tangier disease during an episode of urosepsis. Within two weeks post recovery the dyslilpidemia Tangier disease (familial alpha lipoprotein resolved and the plasma lipid profile returned to deficiency) is a rare genetic disorder of lipid baseline values. metabolism characterized principally by a severe deficiency of high density lipoprotein (HDL), Clinical History accompanied by low plasma levels of low density lipoprotein (LDL) cholesterol, normal or elevated The patient was a 58-year-old woman presenting plasma levels of triglycerides, and accumulation of with the chief complaint of generalized abdominal cholesteryl ester in reticuloendothelial tissue [1, 2]. pain with nausea, vomiting, and fever for four Phenotypically, patients with Tangier disease may days. -

Hypertriglyceridemia
J Am Board Fam Med: first published as 10.3122/jabfm.19.3.310 on 3 May 2006. Downloaded from EVIDENCED-BASED CLINICAL MEDICINE Hypertriglyceridemia Rade N. Pejic, MD, and Daniel T. Lee, MD Hypertriglyceridemia is a commonly encountered lipid abnormality frequently associated with other lipid and metabolic derangements. The National Cholesterol Education Program recommends obtaining a fasting lipid panel in adults over the age of 20. The discovery of hypertriglyceridemia should prompt an investigation for secondary causes such as high fat diet, excessive alcohol intake, certain medica- tions, and medical conditions (eg, diabetes mellitus, hypothyroidism). In addition, patients should be evaluated for other components of the metabolic syndrome. These include abdominal obesity, insulin resistance, low high-density lipoprotein (HDL), high triglyceride, and hypertension. Hypertriglyceride- mia is classified as primary hypertriglyceridemia when there are no secondary causes identified. Pri- mary hypertriglyceridemia is the result of various genetic defects leading to disordered triglyceride me- tabolism. It is important to treat hypertriglyceridemia to prevent pancreatitis by reducing triglyceride levels to <500 mg/dL. Furthermore, lowering triglycerides while treating other dyslipidemias and com- ponents of the metabolic syndrome will reduce coronary events. However, it is controversial how much isolated hypertriglyceridemia correlates directly with coronary artery disease and further studies are needed to clarify whether treatment for this condition leads to meaningful clinical outcomes. Therapeu- tic lifestyle changes (TLC) are the first line of treatment for hypertriglyceridemia. These changes include a low saturated fat, carbohydrate-controlled diet, combined with alcohol reduction, smoking cessation, and regular aerobic exercise. High doses of omega-3 fatty acids from fish and fish oil supplements will lower triglyceride levels significantly. -
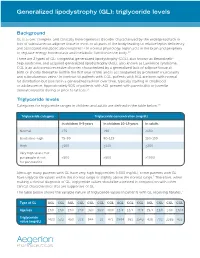
Generalized Lipodystrophy (GL): Triglyceride Levels
Generalized lipodystrophy (GL): triglyceride levels Background GL is a rare, complex, and clinically heterogeneous disorder characterized by the widespread lack or loss of subcutaneous adipose tissue in most or all parts of the body leading to relative leptin deficiency and associated metabolic abnormalities.1,2 In normal physiology, leptin acts in the brain and periphery to regulate energy homeostasis and metabolic function in the body.2,3 There are 2 types of GL: congenital generalized lipodystrophy (CGL), also known as Berardinelli- Seip syndrome, and acquired generalized lipodystrophy (AGL), also known as Lawrence syndrome. CGL is an autosomal recessive disorder characterized by a generalized lack of adipose tissue at birth or shortly thereafter (within the first year of life) and is accompanied by prominent muscularity and subcutaneous veins.1 In contrast to patients with CGL, patients with AGL are born with normal fat distribution but lose fat in a generalized fashion over time, typically starting in childhood or adolescence. Approximately 50% of patients with AGL present with panniculitis or juvenile dermatomyositis during or prior to fat loss.1,4 Triglyceride levels Categories for triglyceride ranges in children and adults are defined in the table below.5,6 Triglyceride category Triglyceride concentration (mg/dL) In children 0-9 years In children 10-19 years In adults Normal <75 <90 <150 Borderline-high 75-99 90-129 150-199 High ≥100 ≥130 ≥200 Very high levels that put people at risk >500 >500 >1000 for pancreatitis Although many patients with GL have very high triglycerides (>500 mg/dL), some patients with GL have triglyceride values within the normal range or slightly above the normal range.7 Therefore, when making a clinical diagnosis of GL, triglyceride values should be assessed in conjunction with other clinical characteristics that are supportive of GL.