Environmental Health Criteria 88 Polychlorinated Dibenzo-Para
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

2,4-Dichlorophenoxyacetic Acid
2,4-Dichlorophenoxyacetic acid 2,4-Dichlorophenoxyacetic acid IUPAC (2,4-dichlorophenoxy)acetic acid name 2,4-D Other hedonal names trinoxol Identifiers CAS [94-75-7] number SMILES OC(COC1=CC=C(Cl)C=C1Cl)=O ChemSpider 1441 ID Properties Molecular C H Cl O formula 8 6 2 3 Molar mass 221.04 g mol−1 Appearance white to yellow powder Melting point 140.5 °C (413.5 K) Boiling 160 °C (0.4 mm Hg) point Solubility in 900 mg/L (25 °C) water Related compounds Related 2,4,5-T, Dichlorprop compounds Except where noted otherwise, data are given for materials in their standard state (at 25 °C, 100 kPa) 2,4-Dichlorophenoxyacetic acid (2,4-D) is a common systemic herbicide used in the control of broadleaf weeds. It is the most widely used herbicide in the world, and the third most commonly used in North America.[1] 2,4-D is also an important synthetic auxin, often used in laboratories for plant research and as a supplement in plant cell culture media such as MS medium. History 2,4-D was developed during World War II by a British team at Rothamsted Experimental Station, under the leadership of Judah Hirsch Quastel, aiming to increase crop yields for a nation at war.[citation needed] When it was commercially released in 1946, it became the first successful selective herbicide and allowed for greatly enhanced weed control in wheat, maize (corn), rice, and similar cereal grass crop, because it only kills dicots, leaving behind monocots. Mechanism of herbicide action 2,4-D is a synthetic auxin, which is a class of plant growth regulators. -

Toxic Residents: Health and Citizenship at Love Canal
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Bucknell University Bucknell University Bucknell Digital Commons Faculty Journal Articles Faculty Scholarship Fall 2016 Toxic Residents: Health and Citizenship at Love Canal Jennifer Thomson Bucknell University, [email protected] Follow this and additional works at: https://digitalcommons.bucknell.edu/fac_journ Part of the Other History Commons, and the United States History Commons Recommended Citation Thomson, Jennifer. "Toxic Residents: Health and Citizenship at Love Canal." Journal of Social History (2016) : 204-223. This Article is brought to you for free and open access by the Faculty Scholarship at Bucknell Digital Commons. It has been accepted for inclusion in Faculty Journal Articles by an authorized administrator of Bucknell Digital Commons. For more information, please contact [email protected]. Journal of Social History Advance Access published December 19, 2015 JENNIFER THOMSON Toxic Residents: Health and Citizenship at Love Canal Abstract Downloaded from This article investigates the relationship between American political culture and grassroots environmentalism in the 1970s. To do so, it examines how the white working class residents of Love Canal, New York, claimed health and a healthy en- vironment as rights of citizenship. To date, the Canal has remained a sore spot for environmental scholarship; this article demonstrates how the analytic difficulties http://jsh.oxfordjournals.org/ posed by the Canal stem from the crosscurrents of American political culture in the late 1970s. Canal residents put their local experience into several larger frames of reference: the rights and responsibilities of citizenship, the plight of Cuban and Vietnamese refugees, and a culture of skepticism toward government and medical authority. -

Approaches for Assessing Health Risks from Complex Mixtures in Indoor Air: a Panel Overview by Carol J
Environmental Health Perspectives Vol. 95, pp. 135-143, 1991 Approaches for Assessing Health Risks from Complex Mixtures in Indoor Air: A Panel Overview by Carol J. Henry,* Lawrence Fishbein,* William J. Meggs,t Nicholas A. Ashford,' Paul A. Schulte,§ Henry Anderson,/' J. Scott Osborne,' and Daniel W. Sepkovictt Critical to a more definitive human health assessment ofthe potential health risks from exposure to complex mixtures in indoor air is the need for a more definitive clinical measure and etiology ofthe helath effects ofcomplex mixtures. This panel overview highlights six ofthe eight presentations ofthe conference panel discussion and features a number ofthe major topical areas of indoor air concern. W. G. Meggs assessed clinical research priorities with primary focus on the role ofvolatile organic chemicals in human health, recognizing the areas where definitive data are lacking. By recogniz- ing many types ofchemical sensitivity, it may be possible to design studies that can illuminate the mechanisms by which chemical exposure may cause disease. The critically important topic of multiple chemical sensitivity was discussed by N. A. Ashford, who identified four high risk groups and defined the demographics ofthese groups. P. A. Schulte address- ed the issue ofbiological markers ofsusceptibility with specific considerations ofboth methodologia and societal aspects that may be operative in the ability to detect innate or inborne differences between individuals and populations. Three case studies were reviewed. H. Anderson discussed the past and present priorities from a public health perspective, focusing on those issues dealing with exposures to environmental tobacco smoke and formaldehyde off-gassing from materials used in mobile homeconstruction. -

Victims of Seveso Disaster Face Higher Risk from Some Cancers 14 September 2009
Victims of Seveso disaster face higher risk from some cancers 14 September 2009 People living in the Seveso area of Italy, which was excess of lymphatic and hematopoietic risk. We did exposed to dioxin after an industrial accident in not identify an all-cancer excess, as seen in 1976, have experienced an increased risk of occupational cohorts which had similar, sometimes developing cancer. Researchers writing in BioMed higher, and more complex exposures". Central's open access journal Environmental Health found an increased risk of breast cancer in More information: Cancer incidence in the women from the most exposed zone and an population exposed to dioxin after the "Seveso excess of lymphatic and hematopoietic tissue accident": twenty years of follow-up; Angela Cecilia neoplasms in all but the least exposed zone. Pesatori, Dario Consonni, Maurizia Rubagotti, Paolo Grillo and Pier Alberto Bertazzi; Angela Pesatori led a team of researchers from the Environmental Health (in press); Fondazione IRCCS Ospedale Maggiore Policlinico, www.ehjournal.net/ a local hospital associated with the University of Milan, who extended a study of cancer incidence in Source: BioMed Central the area, which now covers the period 1977-96. She said, "The industrial accident that occurred in the Seveso area in 1976 exposed a large residential population to substantial amounts of TCDD [2,3,7,8-tetrachlorodibenzo-p-dioxin]. Although the International Agency for Research on Cancer and the US Environmental Protection Agency have both classified TCDD as human carcinogen, scientific debate still persists on the actual cancer risk posed to the general population. We've found that it does pose a carcinogenic hazard, although lower than anticipated from animal studies, at least at the levels experienced by this population after this accident". -

Love Canal Follow-Up Health Study
Love Canal Follow-up Health Study Prepared by the Division of Environmental Health Assessment Center for Environmental Health New York State Department of Health for the U.S. Department of Health and Human Services Agency for Toxic Substances and Disease Registry In memory of Charlene M. Spampinato and her longstanding dedication to the Love Canal project. October 2008 State of New York Funding Provided by Department of Health United States Agency for Toxic Substances and Disease Registry CONTENTS Page LIST OF ABBREVIATIONS………………………………………………....................iii LIST OF FIGURES.……………………………………………………………...............iv LIST OF TABLES ……………………………………………………………….............v LIST OF APPENDICES....…...…...…..…………………………………………...........vii EXECUTIVE SUMMARY………………………………………………………….........1 INTRODUCTION………………………………………………………………………..5 Love Canal History……………………………………………………….6 Environmental Sampling………………………………………………….8 Study Area………………………………………………….....................10 Review of Earlier Love Canal Health Studies…………………………...11 Community Involvement………………………………………………...15 Study Objectives…………………………………………………………17 METHODS………………………………………………………………………………19 Study Population……………………………………………………........19 Comparison Populations………………………………………………....20 Tracing Former Love Canal Residents…………………………………..20 Exposure Assessment………………………………………………........22 Outcome Assessment……………………………………………….…....25 Mortality…………………………………………………….…...25 Cancer Incidence………………………………………………....26 Reproductive Outcomes……………………………………….…28 Potential Confounders…………………………………………………....31 -

Indoor Air Quality Factors in Designing a Healthy Building,” Annual
Spengler, J.D. and Chen, Q. 2000. “Indoor air quality factors in designing a healthy building,” Annual Review of Energy and the Environment, 25, 567-600. Indoor Air Quality Factors in Designing a Healthy Building John D. Spengler School of Public Health, Harvard University, 665 Huntington Avenue, Boston, Massachusetts 02115; e-mail: [email protected] Qingyan (Yan) Chen Building Technology Program, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139; e-mail: [email protected] KEY WORDS: regulations, contaminant sources, building materials and systems, ventilation models, design tools Shortened title: IAQ in Designing a Healthy Building Abstract Current guidelines for green buildings are cursory and inadequate for specifying materials and designing ventilation systems to ensure a healthful indoor environment, i.e. a “healthy building,” by design. Public perception, cultural preferences, litigation trends, current codes and regulations, rapid introduction of new building materials and commercial products, as well as the prevailing design-build practices, pose challenges to systems integration in the design, construction and operation phases of modern buildings. We are on the verge of a paradigm shift in ventilation design thinking. In the past, thermal properties of air within a zone determined heating, ventilating, and air- conditioning (HVAC) specifications. In the future, occupant-specific and highly responsive systems will become the norm. Natural ventilation, displacement ventilation, microzoning with subfloor plenums, along with the use of point of source heat control and point of use sensors, will evolve to create a "smart" responsive ventilation-building dynamic system. Advanced ventilation design tools such as the modeling of computational fluid dynamics (CFD) will be used routinely. -

Polychlorinated Dibenzo-P-Dioxins and Dibenzofurans in the Great Lakes
Hdb Env Chem Vol. 5, Part N (2006): 71–150 DOI 10.1007/698_5_040 © Springer-Verlag Berlin Heidelberg 2005 Published online: 2 December 2005 Polychlorinated Dibenzo-p-dioxins and Dibenzofurans in the Great Lakes Ross J. Norstrom Centre for Analytical and Environmental Chemistry, Department of Chemistry, Carleton University, Ottawa, ON K1S 5B6, Canada [email protected] 1Introduction................................... 73 2 Occurrence and Geographical Distribution .................. 75 2.1 Air........................................ 75 2.1.1ConcentrationsinAir.............................. 75 2.1.2AirDepositionModels............................. 78 2.2 Water....................................... 83 2.2.1SaginawRiver.................................. 83 2.2.2DetroitRiver................................... 84 2.2.3NiagaraRiver.................................. 85 2.3 LakeSediments................................. 86 2.4 RiverandBaySediments............................ 91 2.4.1 Fox River/GreenBaySediments........................ 91 2.4.2LakeSuperiorBaySediments.......................... 91 2.4.3 Saginaw River/SaginawBaySediments.................... 92 2.4.4DetroitRiverSediments............................. 94 2.4.5NiagaraRiverSediments............................ 95 2.5 Fish........................................ 97 2.5.1 Surveys of 2378-TeCDD and 2378-TeCDF . ................ 98 2.5.2ComprehensiveSurveys............................. 103 2.6 SeabirdsandSnappingTurtleEggs...................... 107 2.6.1HerringGullEggs............................... -

History of Seed in the U.S. the Untold American Revolution 660 Pennsylvania Ave SE Suite 302 Washington, D.C
History of Seed in the U.S. The Untold American Revolution 660 Pennsylvania Ave SE Suite 302 Washington, D.C. 20003 P (202) 547-9359 F (202) 547-9429 www.centerforfoodsafety.org Save Our Seeds An exhibition at the National Archives in Washington, D.C., What’s Cooking Uncle Sam?, traces the history of U.S. agriculture from “the horse and plow (SOS) to today’s mechanized farm.” While the exhibition contains humorous elements, including a corporate campaign to win the War Food A program of the Administration’s endorsement of its Vitamin Donuts—“For pep and vigor… Center for Food Safety Vitamin Donuts!”—it also chronicles a sobering story of American farming and how the effects of U.S. food and agricultural policies reach far beyond the borders of Uncle Sam. Throughout, it is clear that the path of agriculture begins with the seed. Over the past 40 years, the U.S. has led a radical shift toward commercialization, consolidation, and control of seed. Prior to the advent of industrial agriculture, there were thousands of seed companies and public breeding institutions. At present, the top 10 seed and chemical companies, with the majority stake owned by U.S. corporations, control 73 1 Debbie Barker percent of the global market. International Program Today, fewer than 2 percent of Americans are farmers,2 whereas 90 percent 3 Director of our citizens lived on farms in 1810. This represents perhaps a more transformative revolution than even the Revolutionary War recorded in our history books. August 2012 This report will provide a summary of U.S. -

Congener Patterns of Polychlorinated Dibenzo-P-Dioxins
Science of the Total Environment 746 (2020) 141098 Contents lists available at ScienceDirect Science of the Total Environment journal homepage: www.elsevier.com/locate/scitotenv Review Congener patterns of polychlorinated dibenzo-p-dioxins, dibenzofurans and biphenyls as a useful aid to source identification during a contamination incident in the food chain Ron L.A.P. Hoogenboom a,⁎, Rainer Malisch b, Stefan P.J. van Leeuwen a, Huig Vanderperren c, Helge Hove d, Alwyn Fernandes f, Alexander Schächtele b, Martin Rose e,g a Wageningen Food Safety Research, Wageningen UR, Akkermaalsbos 2, 6708 WB Wageningen, the Netherlands b EURL for POPs, CVUA, Bissierstraße 5, 79114 Freiburg, Germany c FLVV, Leuvensesteenweg 17, 3080 Tervuren, Belgium d NIFES, Strandgaten 229, 5004 Bergen, Norway e FERA Science Ltd, Sand Hutton, York YO41 1LZ, UK f School of Environmental Sciences, University of East Anglia, Norwich NR4 7TJ, UK g Manchester Institute of Biotechnology, University of Manchester, Manchester, UK HIGHLIGHTS GRAPHICAL ABSTRACT • Congener patterns are important tools for source identification. • TEQ contribution may be preferred over contribution to total sum. • Only PCDDs point to clay or chlorophenols, only PCDFs to PCB sources. • Mixed patterns point to minerals or burning processes. • Simple patterns indicate certain pesti- cides or herbicides. article info abstract Article history: Polychlorinated dibenzo-p-dioxins (PCDDs), dibenzofurans (PCDFs) and biphenyls (PCBs) are still consid- Received 7 May 2020 ered among the most important groups of contaminants in the food chain. Self-control by food producers Received in revised form 13 July 2020 and official control by authorities are important activities that allow contaminant sources to be traced Accepted 18 July 2020 and promote further reduction in food and feed levels. -
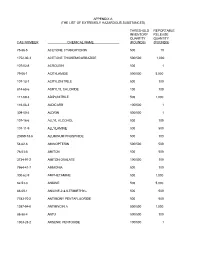
The List of Extremely Hazardous Substances)
APPENDIX A (THE LIST OF EXTREMELY HAZARDOUS SUBSTANCES) THRESHOLD REPORTABLE INVENTORY RELEASE QUANTITY QUANTITY CAS NUMBER CHEMICAL NAME (POUNDS) (POUNDS) 75-86-5 ACETONE CYANOHYDRIN 500 10 1752-30-3 ACETONE THIOSEMICARBAZIDE 500/500 1,000 107-02-8 ACROLEIN 500 1 79-06-1 ACRYLAMIDE 500/500 5,000 107-13-1 ACRYLONITRILE 500 100 814-68-6 ACRYLYL CHLORIDE 100 100 111-69-3 ADIPONITRILE 500 1,000 116-06-3 ALDICARB 100/500 1 309-00-2 ALDRIN 500/500 1 107-18-6 ALLYL ALCOHOL 500 100 107-11-9 ALLYLAMINE 500 500 20859-73-8 ALUMINUM PHOSPHIDE 500 100 54-62-6 AMINOPTERIN 500/500 500 78-53-5 AMITON 500 500 3734-97-2 AMITON OXALATE 100/500 100 7664-41-7 AMMONIA 500 100 300-62-9 AMPHETAMINE 500 1,000 62-53-3 ANILINE 500 5,000 88-05-1 ANILINE,2,4,6-TRIMETHYL- 500 500 7783-70-2 ANTIMONY PENTAFLUORIDE 500 500 1397-94-0 ANTIMYCIN A 500/500 1,000 86-88-4 ANTU 500/500 100 1303-28-2 ARSENIC PENTOXIDE 100/500 1 THRESHOLD REPORTABLE INVENTORY RELEASE QUANTITY QUANTITY CAS NUMBER CHEMICAL NAME (POUNDS) (POUNDS) 1327-53-3 ARSENOUS OXIDE 100/500 1 7784-34-1 ARSENOUS TRICHLORIDE 500 1 7784-42-1 ARSINE 100 100 2642-71-9 AZINPHOS-ETHYL 100/500 100 86-50-0 AZINPHOS-METHYL 10/500 1 98-87-3 BENZAL CHLORIDE 500 5,000 98-16-8 BENZENAMINE, 3-(TRIFLUOROMETHYL)- 500 500 100-14-1 BENZENE, 1-(CHLOROMETHYL)-4-NITRO- 500/500 500 98-05-5 BENZENEARSONIC ACID 10/500 10 3615-21-2 BENZIMIDAZOLE, 4,5-DICHLORO-2-(TRI- 500/500 500 FLUOROMETHYL)- 98-07-7 BENZOTRICHLORIDE 100 10 100-44-7 BENZYL CHLORIDE 500 100 140-29-4 BENZYL CYANIDE 500 500 15271-41-7 BICYCLO[2.2.1]HEPTANE-2-CARBONITRILE,5- -

The Seveso Disaster Legacy Laura Centemeri
The Seveso disaster legacy Laura Centemeri To cite this version: Laura Centemeri. The Seveso disaster legacy. Nature and History in Modern Italy, Ohio University Press & Swallow Press, pp.251-273, 2010. hal-01016045 HAL Id: hal-01016045 https://hal.archives-ouvertes.fr/hal-01016045 Submitted on 30 Jul 2014 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. 233 The Seveso Disaster Legacy Laura Centemeri 1. Introduction In the history of the environment as a public problem, industrial disasters have been insufficiently explored.1 Such disasters are nonetheless crucial because their collective interpretation weaves technical and scientific issues with problems of social justice and controversies concerning conflicting “common goods”, by destabilizing the equilibria that has formed between these elements.2 These disequilibria open the way for episodes of normative and cognitive uncertainty, and thereby become windows of opportunity for social critique and social change, especially by opening public debates on rules, institutions, and representations about technical progress. In short, industrial disasters become opportunities for rethinking the types of "compromise" between "orders of worth"—in particular industrial and civic--upon which a society rests.3 Yet there is still little acknowledgment of the social change that industrial disasters can trigger. -

Environmental Health Criteria 39 PARAQUAT and DIQUAT
Environmental Health Criteria 39 PARAQUAT AND DIQUAT Please note that the layout and pagination of this web version are not identical with the printed version. Paraquat and diquat (EHC 39, 1984) INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY ENVIRONMENTAL HEALTH CRITERIA 39 PARAQUAT AND DIQUAT This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organisation, or the World Health Organization. Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization World Health Orgnization Geneva, 1984 The International Programme on Chemical Safety (IPCS) is a joint venture of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization. The main objective of the IPCS is to carry out and disseminate evaluations of the effects of chemicals on human health and the quality of the environment. Supporting activities include the development of epidemiological, experimental laboratory, and risk-assessment methods that could produce internationally comparable results, and the development of manpower in the field of toxicology. Other activities carried out by the IPCS include the development of know-how for coping with chemical accidents, coordination of laboratory testing and epidemiological studies, and promotion of research on the mechanisms of the biological action of chemicals. ISBN 92 4 154099 4 The World Health Organization welcomes requests for permission to reproduce or translate its publications, in part or in full. Applications and enquiries should be addressed to the Office of Publications, World Health Organization, Geneva, Switzerland, which will be glad to provide the latest information on any changes made to the text, plans for new editions, and reprints and translations already available.