Cnvs Leading to Fusion Transcripts in Individuals with Autism Spectrum Disorder
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Constitutive Activation of RAS/MAPK Pathway Cooperates with Trisomy 21 and Is Therapeutically Exploitable in Down Syndrome B-Cell Leukemia
Author Manuscript Published OnlineFirst on March 27, 2020; DOI: 10.1158/1078-0432.CCR-19-3519 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Constitutive activation of RAS/MAPK pathway cooperates with trisomy 21 and is therapeutically exploitable in Down syndrome B-cell Leukemia Anouchka P. Laurent1,2, Aurélie Siret1, Cathy Ignacimouttou1, Kunjal Panchal3, M’Boyba K. Diop4, Silvia Jenny5, Yi-Chien Tsai5, Damien Ross-Weil1, Zakia Aid1, Naïs Prade6, Stéphanie Lagarde6, Damien Plassard7, Gaelle Pierron8, Estelle Daudigeos-Dubus4, Yann Lecluse4, Nathalie Droin1, Beat Bornhauser5, Laurence C. Cheung3,9, John D. Crispino10, Muriel Gaudry1, Olivier A. Bernard1, Elizabeth Macintyre11, Carole Barin Bonnigal12, Rishi S. Kotecha3,9,13, Birgit Geoerger4, Paola Ballerini14, Jean-Pierre Bourquin5, Eric Delabesse6, Thomas Mercher1,15 and Sébastien Malinge1,3 1INSERM U1170, Gustave Roussy Institute, Université Paris Saclay, Villejuif, France 2Université Paris Diderot, Paris, France 3Telethon Kids Cancer Centre, Telethon Kids Institute, University of Western Australia, Perth, Australia 4Gustave Roussy Institute Cancer Campus, Department of Pediatric and Adolescent Oncology, INSERM U1015, Equipe Labellisée Ligue Nationale contre le Cancer, Université Paris-Saclay, Villejuif, France 5Department of Pediatric Oncology, Children’s Research Centre, University Children’s Hospital Zurich, Zurich, Switzerland 6Centre of Research on Cancer of Toulouse (CRCT), CHU Toulouse, Université Toulouse III, Toulouse, France 7IGBMC, Plateforme GenomEast, UMR7104 CNRS, Ilkirch, France 8Service de Génétique, Institut Curie, Paris, France 9School of Pharmacy and Biomedical Sciences, Curtin University, Bentley, Australia 10Division of Hematology/Oncology, Northwestern University, Chicago, USA 11Hematology, Université de Paris, Institut Necker-Enfants Malades and Assistance Publique – Hopitaux de Paris, Paris, France 12Centre Hospitalier Universitaire de Tours, Tours, France 1 Downloaded from clincancerres.aacrjournals.org on September 30, 2021. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Primate Specific Retrotransposons, Svas, in the Evolution of Networks That Alter Brain Function
Title: Primate specific retrotransposons, SVAs, in the evolution of networks that alter brain function. Olga Vasieva1*, Sultan Cetiner1, Abigail Savage2, Gerald G. Schumann3, Vivien J Bubb2, John P Quinn2*, 1 Institute of Integrative Biology, University of Liverpool, Liverpool, L69 7ZB, U.K 2 Department of Molecular and Clinical Pharmacology, Institute of Translational Medicine, The University of Liverpool, Liverpool L69 3BX, UK 3 Division of Medical Biotechnology, Paul-Ehrlich-Institut, Langen, D-63225 Germany *. Corresponding author Olga Vasieva: Institute of Integrative Biology, Department of Comparative genomics, University of Liverpool, Liverpool, L69 7ZB, [email protected] ; Tel: (+44) 151 795 4456; FAX:(+44) 151 795 4406 John Quinn: Department of Molecular and Clinical Pharmacology, Institute of Translational Medicine, The University of Liverpool, Liverpool L69 3BX, UK, [email protected]; Tel: (+44) 151 794 5498. Key words: SVA, trans-mobilisation, behaviour, brain, evolution, psychiatric disorders 1 Abstract The hominid-specific non-LTR retrotransposon termed SINE–VNTR–Alu (SVA) is the youngest of the transposable elements in the human genome. The propagation of the most ancient SVA type A took place about 13.5 Myrs ago, and the youngest SVA types appeared in the human genome after the chimpanzee divergence. Functional enrichment analysis of genes associated with SVA insertions demonstrated their strong link to multiple ontological categories attributed to brain function and the disorders. SVA types that expanded their presence in the human genome at different stages of hominoid life history were also associated with progressively evolving behavioural features that indicated a potential impact of SVA propagation on a cognitive ability of a modern human. -

Essential Genes and Their Role in Autism Spectrum Disorder
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2017 Essential Genes And Their Role In Autism Spectrum Disorder Xiao Ji University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Bioinformatics Commons, and the Genetics Commons Recommended Citation Ji, Xiao, "Essential Genes And Their Role In Autism Spectrum Disorder" (2017). Publicly Accessible Penn Dissertations. 2369. https://repository.upenn.edu/edissertations/2369 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/2369 For more information, please contact [email protected]. Essential Genes And Their Role In Autism Spectrum Disorder Abstract Essential genes (EGs) play central roles in fundamental cellular processes and are required for the survival of an organism. EGs are enriched for human disease genes and are under strong purifying selection. This intolerance to deleterious mutations, commonly observed haploinsufficiency and the importance of EGs in pre- and postnatal development suggests a possible cumulative effect of deleterious variants in EGs on complex neurodevelopmental disorders. Autism spectrum disorder (ASD) is a heterogeneous, highly heritable neurodevelopmental syndrome characterized by impaired social interaction, communication and repetitive behavior. More and more genetic evidence points to a polygenic model of ASD and it is estimated that hundreds of genes contribute to ASD. The central question addressed in this dissertation is whether genes with a strong effect on survival and fitness (i.e. EGs) play a specific oler in ASD risk. I compiled a comprehensive catalog of 3,915 mammalian EGs by combining human orthologs of lethal genes in knockout mice and genes responsible for cell-based essentiality. -
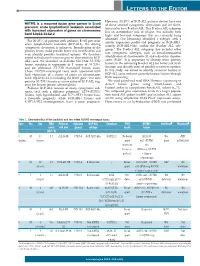
NUTM1 Is a Recurrent Fusion Gene Partner in B-Cell Precursor Acute
LETTERS TO THE EDITOR However, 20-25% of BCP-ALL patients do not have one NUTM1 is a recurrent fusion gene partner in B-cell of these sentinel cytogenetic aberrations and are there- precursor acute lymphoblastic leukemia associated fore said to have B-other ALL. This B-other ALL subgroup with increased expression of genes on chromosome has an intermediate risk of relapse, but includes both band 10p12.31-12.2 high- and low-risk subgroups that are currently being identified. Our laboratory identified a subtype with a For 20-25% of patients with pediatric B-cell precursor similar expression profile and prognosis as BCR-ABL1, acute lymphoblastic leukemia (BCP-ALL), the driving namely BCR-ABL1-like, within the B-other ALL sub- cytogenetic aberration is unknown. Identification of the group.2 The B-other ALL subgroup also includes other primary lesion could provide better risk stratification and rare cytogenetic subtypes, such as intrachromosomal even identify possible treatment options. We therefore amplification of chromosome 21 and a dicentric chromo- aimed to find novel recurrent genetic aberrations in BCP- 1 ALL cases. We identified an in-frame SLC12A6-NUTM1 some (9;20). It is important to identify more primary fusion, resulting in expression of 3’ exons of NUTM1, lesions in the remaining B-other ALL for better risk strat- and six additional NUTM1-rearranged fusion cases. ification and identification of possible treatment options. These NUTM1-rearranged cases were associated with In this study, we aimed to identify recurrent fusions in high expression of a cluster of genes on chromosome BCP-ALL cases without currently known lesions through band 10p12.31-12.2, including the BMI1 gene. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Transposable Elements in Human Cancers by Genome-Wide EST Alignment
Genes Genet. Syst. (2007) 82, p. 145–156 Transposable elements in human cancers by genome-wide EST alignment Dae-Soo Kim1, Jae-Won Huh2 and Heui-Soo Kim1,2* 1PBBRC, Interdisciplinary Research Program of Bioinformatics, Pusan National University, Busan 609-735, Republic of Korea 2Division of Biological Sciences, College of Natural Sciences, Pusan National University, Busan 609-735, Republic of Korea (Received 24 November 2006, accepted 23 January 2007) Transposable elements may affect coding sequences, splicing patterns, and tran- scriptional regulation of human genes. Particles of the transposable elements have been detected in several tissues and tumors. Here, we report genome-wide analysis of gene expression regulated by transposable elements in human cancers. We adopted an analysis pipeline for screening methods to detect cancer- specific expression from expressed human sequences. We developed a database (TECESdb) for understanding the mechanism of cancer development in relation to transposable elements. A total of 999 genes fused with transposable elements were found to be cancer-related in our analysis of the EST database. According to GO (Gene Ontology) analysis, the majority of the 999 cancer-specific genes have functional association with gene receptor, DNA binding, and kinase activity. Our data could contribute greatly to our understanding of human cancers in relation to transposable elements. Key words: Transposable elements, Cancer, Fusion gene, Bioinformatics, EST also appeared in open-reading frames of functional INTRODUCTION human genes (Yulug et al., 1995; Makalowski et al., 1999; The human genome is estimated to be composed of 45% Nekrutenko and Li, 2001; Huh et al., 2006). transposable elements (International Human Genome The L1 5’UTR element is known to have an antisense Sequencing Consortium 2001). -

"The Genecards Suite: from Gene Data Mining to Disease Genome Sequence Analyses". In: Current Protocols in Bioinformat
The GeneCards Suite: From Gene Data UNIT 1.30 Mining to Disease Genome Sequence Analyses Gil Stelzer,1,5 Naomi Rosen,1,5 Inbar Plaschkes,1,2 Shahar Zimmerman,1 Michal Twik,1 Simon Fishilevich,1 Tsippi Iny Stein,1 Ron Nudel,1 Iris Lieder,2 Yaron Mazor,2 Sergey Kaplan,2 Dvir Dahary,2,4 David Warshawsky,3 Yaron Guan-Golan,3 Asher Kohn,3 Noa Rappaport,1 Marilyn Safran,1 and Doron Lancet1,6 1Department of Molecular Genetics, Weizmann Institute of Science, Rehovot, Israel 2LifeMap Sciences Ltd., Tel Aviv, Israel 3LifeMap Sciences Inc., Marshfield, Massachusetts 4Toldot Genetics Ltd., Hod Hasharon, Israel 5These authors contributed equally to the paper 6Corresponding author GeneCards, the human gene compendium, enables researchers to effectively navigate and inter-relate the wide universe of human genes, diseases, variants, proteins, cells, and biological pathways. Our recently launched Version 4 has a revamped infrastructure facilitating faster data updates, better-targeted data queries, and friendlier user experience. It also provides a stronger foundation for the GeneCards suite of companion databases and analysis tools. Improved data unification includes gene-disease links via MalaCards and merged biological pathways via PathCards, as well as drug information and proteome expression. VarElect, another suite member, is a phenotype prioritizer for next-generation sequencing, leveraging the GeneCards and MalaCards knowledgebase. It au- tomatically infers direct and indirect scored associations between hundreds or even thousands of variant-containing genes and disease phenotype terms. Var- Elect’s capabilities, either independently or within TGex, our comprehensive variant analysis pipeline, help prepare for the challenge of clinical projects that involve thousands of exome/genome NGS analyses. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

EWSR1 Gene EWS RNA Binding Protein 1
EWSR1 gene EWS RNA binding protein 1 Normal Function The EWSR1 gene provides instructions for making the EWS protein, whose function is not completely understood. The EWS protein has two regions that contribute to its function. One region, the transcriptional activation domain, allows the EWS protein to turn on (activate) the first step in the production of proteins from genes (transcription). The other region, the RNA-binding domain, allows the EWS protein to attach (bind) to the genetic blueprint for proteins called RNA. The EWS protein may be involved in piecing together this blueprint. Some studies suggest that the RNA-binding domain is able to block (inhibit) the activity of the transcriptional activation domain, and thus regulate the function of the EWS protein. Health Conditions Related to Genetic Changes Ewing sarcoma Mutations involving the EWSR1 gene can cause a type of cancerous tumor known as Ewing sarcoma. These tumors develop in bones or soft tissues, such as nerves and cartilage. There are several types of Ewing sarcoma, including Ewing sarcoma of bone, extraosseous Ewing sarcoma, peripheral primitive neuroectodermal tumor, and Askin tumor. The mutations that cause these tumors are acquired during a person's lifetime and are present only in the tumor cells. This type of genetic change, called a somatic mutation, is not inherited. The most common mutation that causes Ewing sarcoma is a rearrangement (translocation) of genetic material between chromosome 22 and chromosome 11. This translocation, written as t(11;22), fuses part of the EWSR1 gene on chromosome 22 with part of another gene on chromosome 11 called FLI1, creating an EWSR1/FLI1 fusion gene. -

Birth of a Chimeric Primate Gene by Capture of the Transposase Gene
Birth of a chimeric primate gene by capture of the SEE COMMENTARY transposase gene from a mobile element Richard Cordaux*, Swalpa Udit†, Mark A. Batzer*, and Ce´ dric Feschotte†‡ *Department of Biological Sciences, Biological Computation and Visualization Center, Center for BioModular Multi-Scale Systems, Louisiana State University, 202 Life Sciences Building, Baton Rouge, LA 70803; and †Department of Biology, University of Texas, Arlington, TX 76019 Edited by Susan R. Wessler, University of Georgia, Athens, GA, and approved March 27, 2006 (received for review February 10, 2006) The emergence of new genes and functions is of central impor- SETMAR transcript, which consists of these three exons, is tance to the evolution of species. The contribution of various types predicted to encode a protein of 671 amino acids and is of duplications to genetic innovation has been extensively inves- supported by 48 human cDNA clones from 18 different normal tigated. Less understood is the creation of new genes by recycling and͞or cancerous tissues (Table 1, which is published as sup- of coding material from selfish mobile genetic elements. To inves- porting information on the PNAS web site; refs. 14 and 15). tigate this process, we reconstructed the evolutionary history of These data suggest that the SETMAR protein is broadly ex- SETMAR, a new primate chimeric gene resulting from fusion of a pressed and has an important, yet unknown, function in human. SET histone methyltransferase gene to the transposase gene of a Recently, it was shown that the SET domain of the SETMAR mobile element. We show that the transposase gene was recruited protein exhibits histone methyltransferase activity (15), as do all as part of SETMAR 40–58 million years ago, after the insertion of known SET domains (16, 17). -

Selective Induction of Leukemia-Associated Fusion Genes by High-Dose Ionizing Radiation1
[CANCER RESEARCH 58. 421-425. February I. 1<W8| Selective Induction of Leukemia-associated Fusion Genes by High-Dose Ionizing Radiation1 Michael W. N. Deininger, Shikha Bose, Joanna Gora-Tybor, Xiu-Hua Yan, John M. Goldman, and Junia V. Melo2 Leukaemia Research Fumi Centre for Adult Leukaemia. Department of Haemah>li>/;\: Royal Postgraduale Medical School, Ducane Road, London W12 ONN, United Kingdom ABSTRACT event involves the acquisition of the genetic abnormality whose "success" in the production of a leukemic phenotype will depend on There is strong clinical and epidemiológica! evidence that ionizing its capacity to impart to the target cell a proliferative and/or survival radiation can cause leukemia by inducing DNA damage. This crucial advantage over its normal neighbors. In molecular terms, the gener initiation event is believed to be the result of random DNA breakage and misrepair, whereas the subsequent steps, promotion and progression, ation of a potentially successful reciprocal chromosomal translocation must rely on mechanisms of selective pressure to provide the expanding requires that: (a) at least two independent DNA DSBs occur, one in leukemic population with its proliferative/renewal advantage. To investi each chromosome partner; (b) the two breaks occur simultaneously, gate the susceptibility of human cells to external agents at the genetic i.e., within the same cell cycle, so that the two ends of one broken recombination stage of leukemogenesis, we subjected two hematopoietic chromosome are available to interact and be ligated (misrepaired) to cell lines, KG1 and III.6(1, to high doses of y-irradiation. The irradiation the respective complementary broken ends of the other chromosome; induced the formation of fusion genes characteristic of leukemia in both and (c) the recombination observes the polarity of the DNA molecule.